

An optimized workflow for single-cell transcriptomics and repertoire profiling of purified lymphocytes from clinical samples
- PMID: 32042039
- PMCID: PMC7010687
- DOI: 10.1038/s41598-020-58939-y
An optimized workflow for single-cell transcriptomics and repertoire profiling of purified lymphocytes from clinical samples
Erratum in
-
Publisher Correction: An optimized workflow for single-cell transcriptomics and repertoire profiling of purified lymphocytes from clinical samples.Sci Rep. 2020 Apr 14;10(1):6321. doi: 10.1038/s41598-020-62066-z. Sci Rep. 2020. PMID: 32286331 Free PMC article.
Abstract
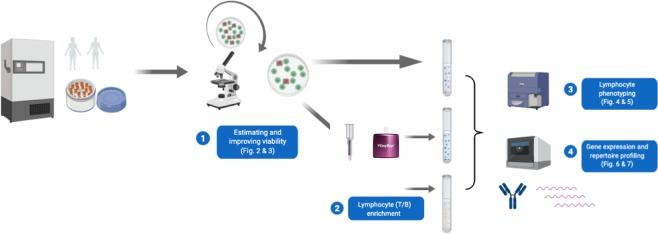
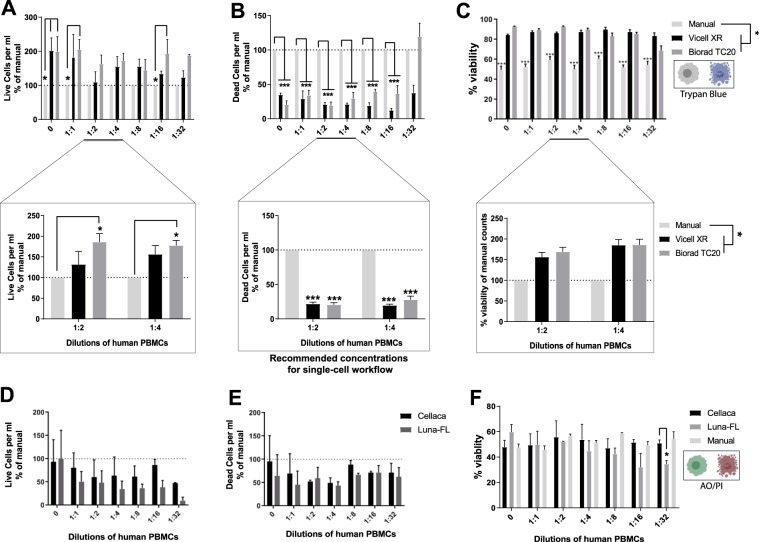
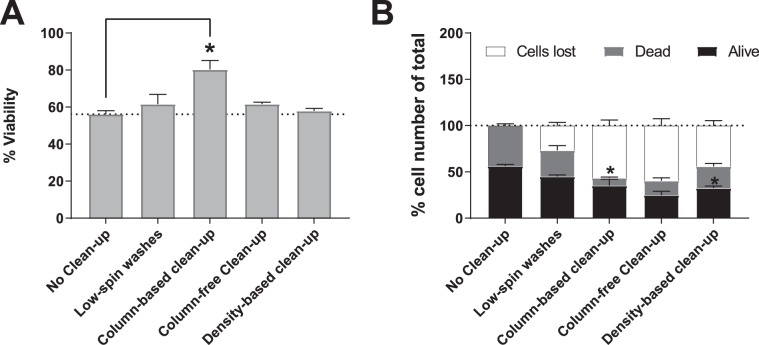
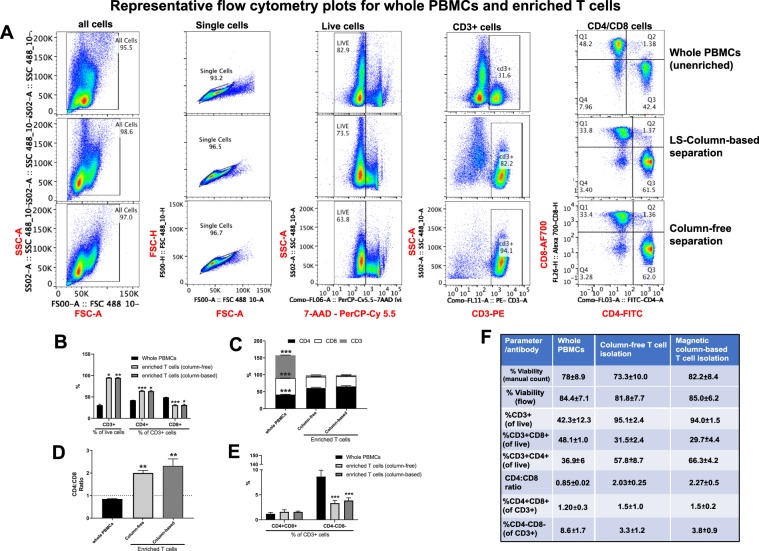
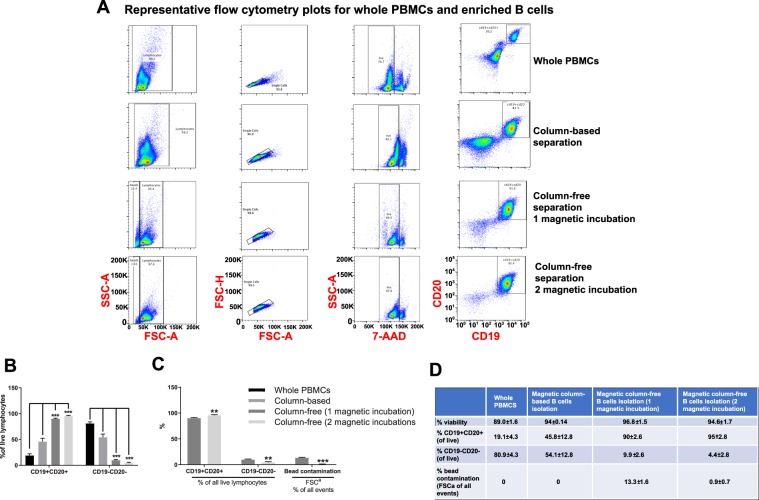
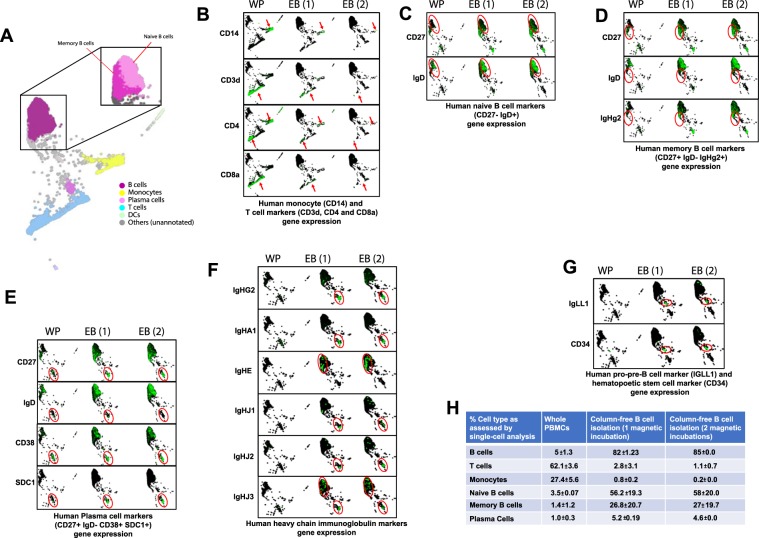
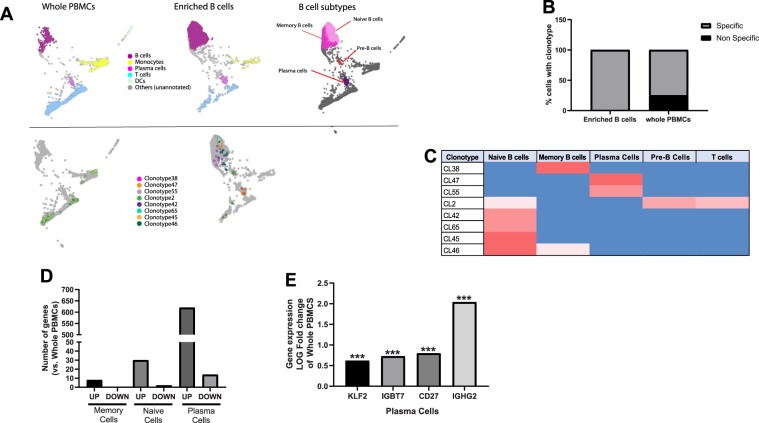
Establishing clinically relevant single-cell (SC) transcriptomic workflows from cryopreserved tissue is essential to move this emerging immune monitoring technology from the bench to the bedside. Improper sample preparation leads to detrimental cascades, resulting in loss of precious time, money and finally compromised data. There is an urgent need to establish protocols specifically designed to overcome the inevitable variations in sample quality resulting from uncontrollable factors in a clinical setting. Here, we explore sample preparation techniques relevant to a range of clinically relevant scenarios, where SC gene expression and repertoire analysis are applied to a cryopreserved sample derived from a small amount of blood, with unknown or partially known preservation history. We compare a total of ten cell-counting, viability-improvement, and lymphocyte-enrichment methods to highlight a number of unexpected findings. Trypan blue-based automated counters, typically recommended for single-cell sample quantitation, consistently overestimate viability. Advanced sample clean-up procedures significantly impact total cell yield, while only modestly increasing viability. Finally, while pre-enrichment of B cells from whole peripheral blood mononuclear cells (PBMCs) results in the most reliable BCR repertoire data, comparable T-cell enrichment strategies distort the ratio of CD4+ and CD8+ cells. Furthermore, we provide high-resolution analysis of gene expression and clonotype repertoire of different B cell subtypes. Together these observations provide both qualitative and quantitative sample preparation guidelines that increase the chances of obtaining high-quality single-cell transcriptomic and repertoire data from human PBMCs in a variety of clinical settings.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
