

Long-range single-molecule mapping of chromatin accessibility in eukaryotes
- PMID: 32042188
- PMCID: PMC7968351
- DOI: 10.1038/s41592-019-0730-2
Long-range single-molecule mapping of chromatin accessibility in eukaryotes
Abstract
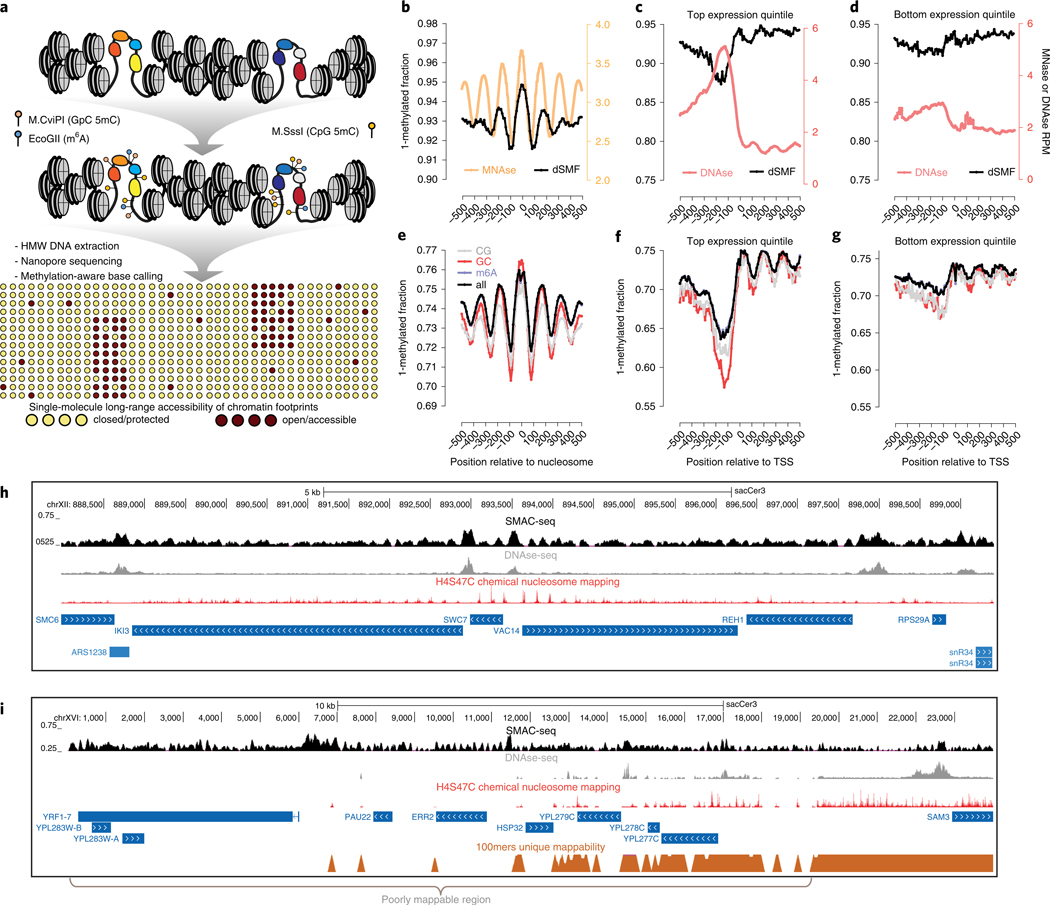
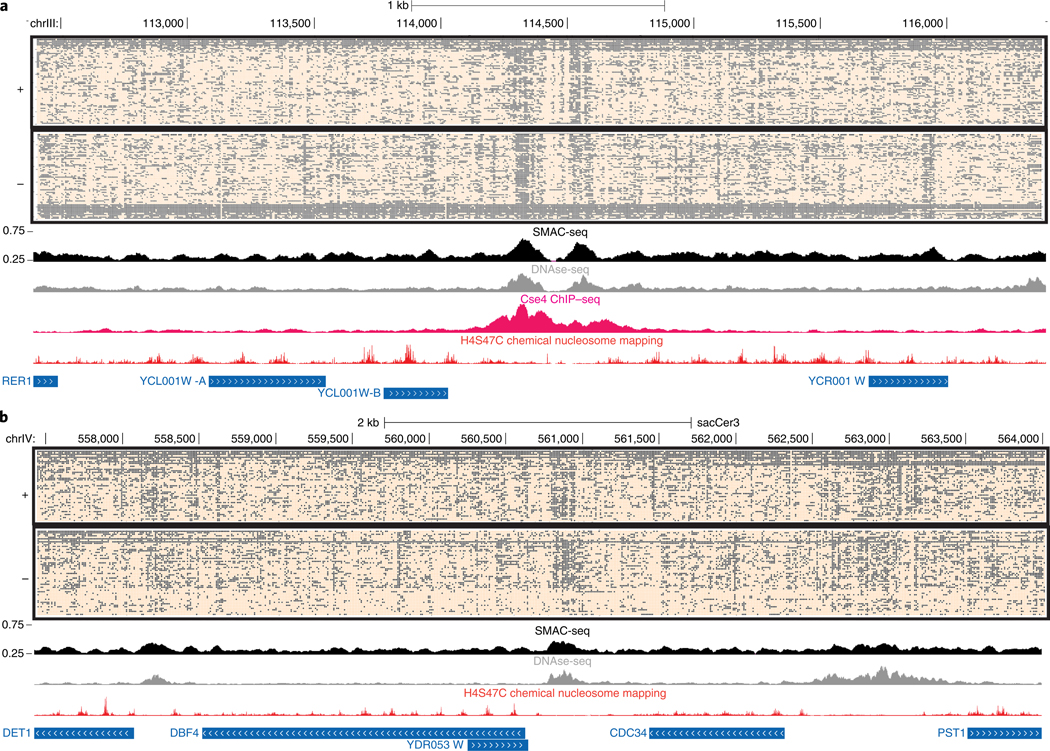
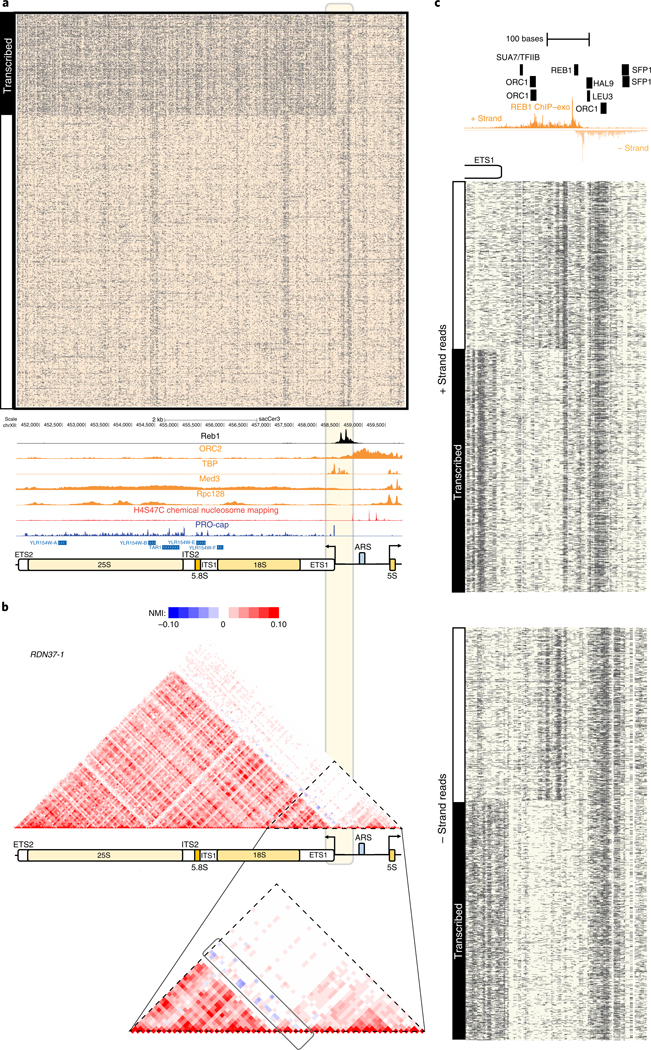
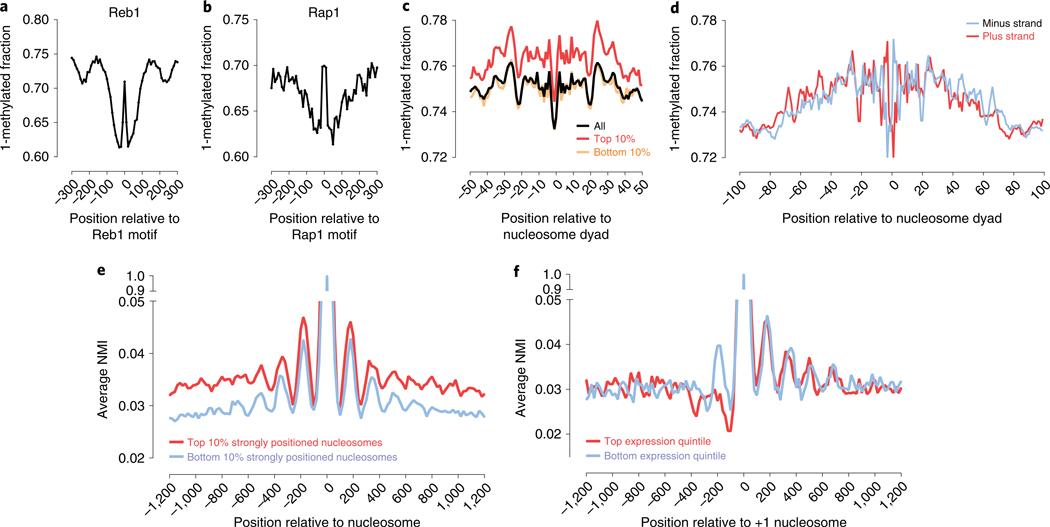
Mapping open chromatin regions has emerged as a widely used tool for identifying active regulatory elements in eukaryotes. However, existing approaches, limited by reliance on DNA fragmentation and short-read sequencing, cannot provide information about large-scale chromatin states or reveal coordination between the states of distal regulatory elements. We have developed a method for profiling the accessibility of individual chromatin fibers, a single-molecule long-read accessible chromatin mapping sequencing assay (SMAC-seq), enabling the simultaneous, high-resolution, single-molecule assessment of chromatin states at multikilobase length scales. Our strategy is based on combining the preferential methylation of open chromatin regions by DNA methyltransferases with low sequence specificity, in this case EcoGII, an N6-methyladenosine (m6A) methyltransferase, and the ability of nanopore sequencing to directly read DNA modifications. We demonstrate that aggregate SMAC-seq signals match bulk-level accessibility measurements, observe single-molecule nucleosome and transcription factor protection footprints, and quantify the correlation between chromatin states of distal genomic elements.
Figures

References
-
- Wu C The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature 286, 854–860 (1980). - PubMed
-
- McGhee JD, Wood WI, Dolan M, Engel JD & Felsenfeld GA 200 base pair region at the 5′ end of the chicken adult β-globin gene is accessible to nuclease digestion. Cell 27, 45–55 (1981). - PubMed
-
- Dorschner MO et al. High-throughput localization of functional elements by quantitative chromatin profiling. Nat. Methods 1, 219–225 (2004). - PubMed
-
- Sabo PJ et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat. Methods 3, 511–518 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
