

Regenerative lineages and immune-mediated pruning in lung cancer metastasis
- PMID: 32042191
- PMCID: PMC7021003
- DOI: 10.1038/s41591-019-0750-6
Regenerative lineages and immune-mediated pruning in lung cancer metastasis
Abstract
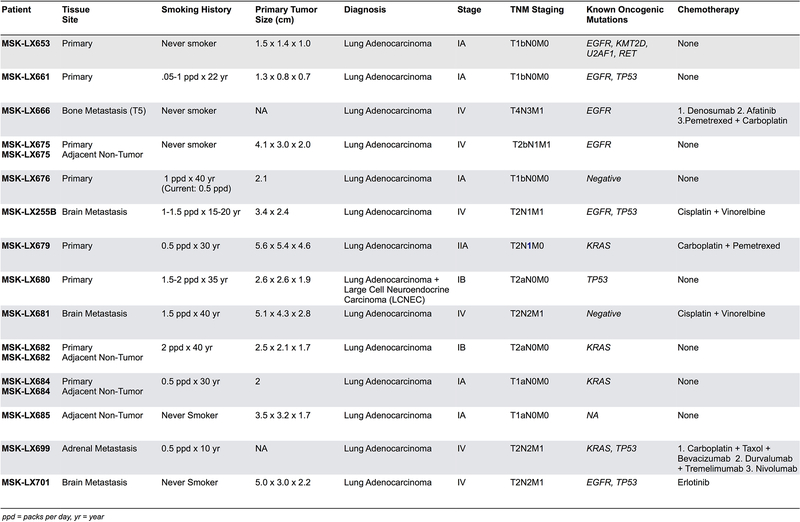
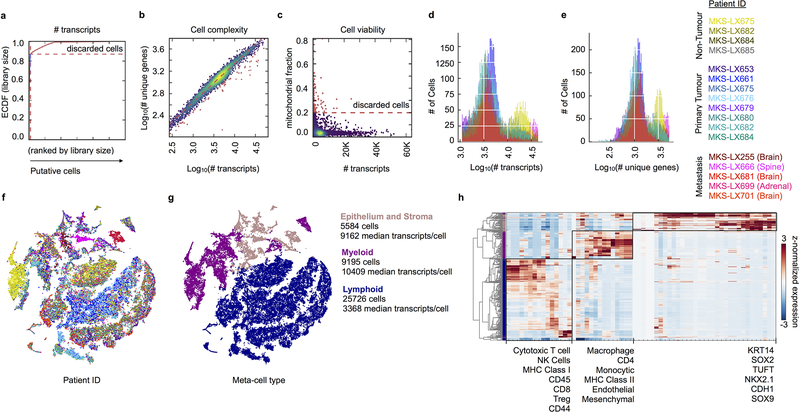
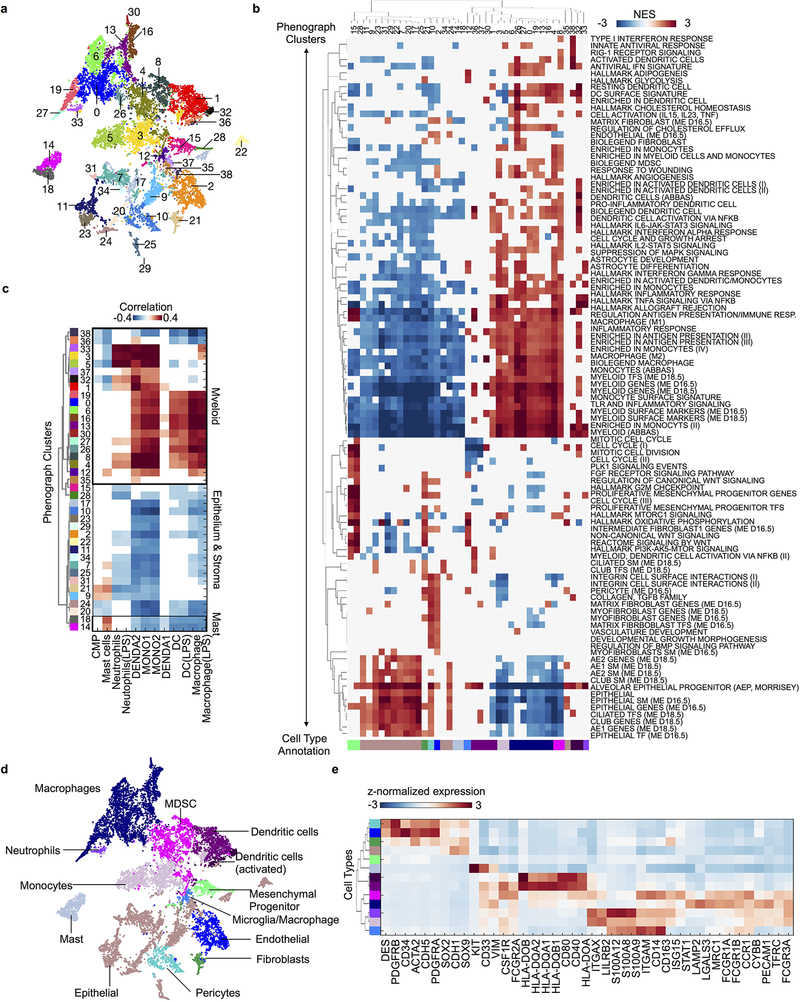
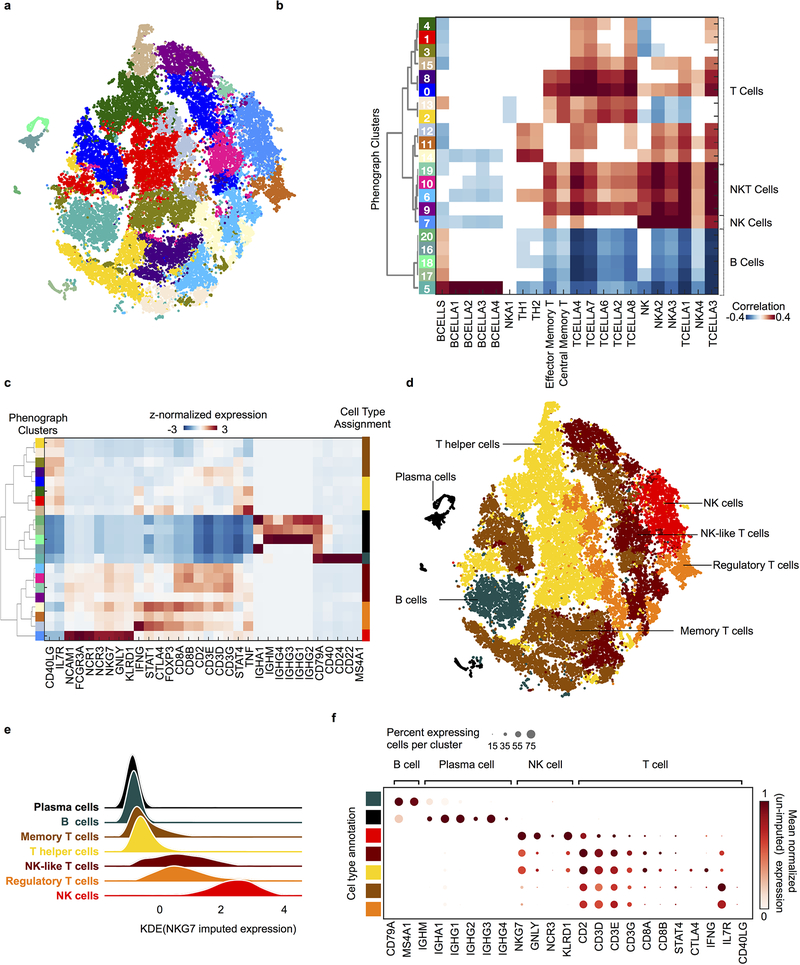
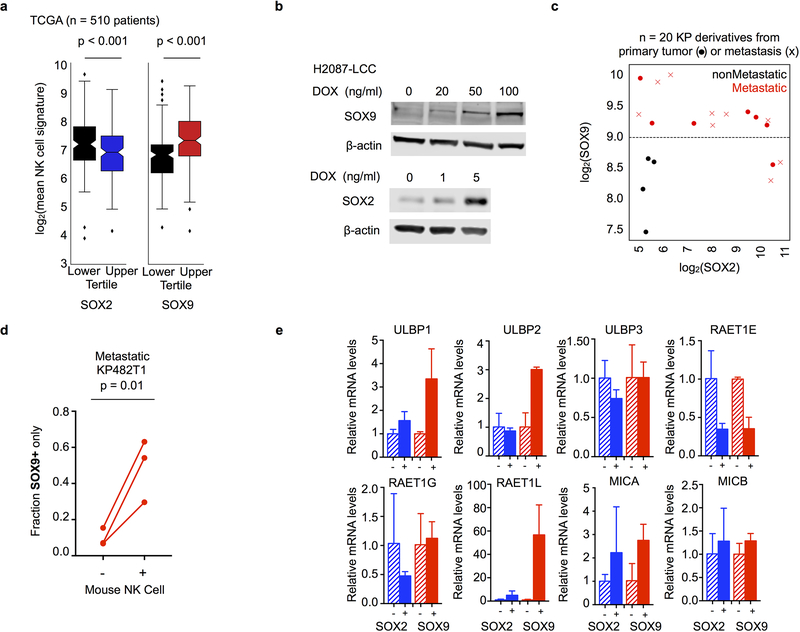
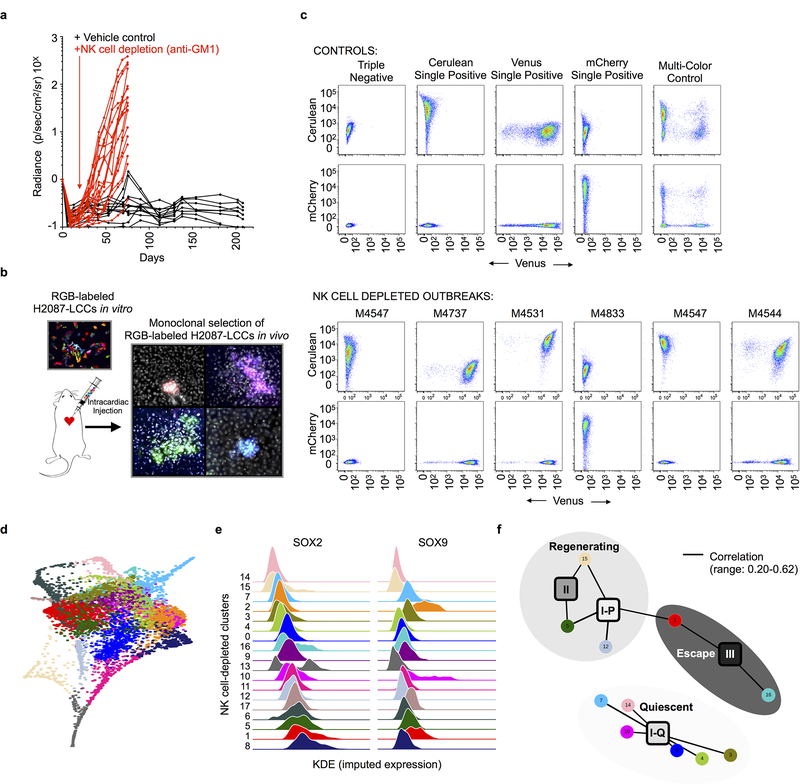
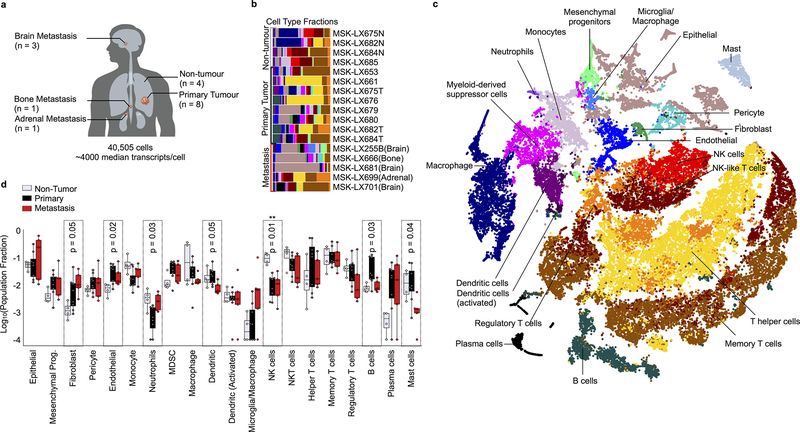
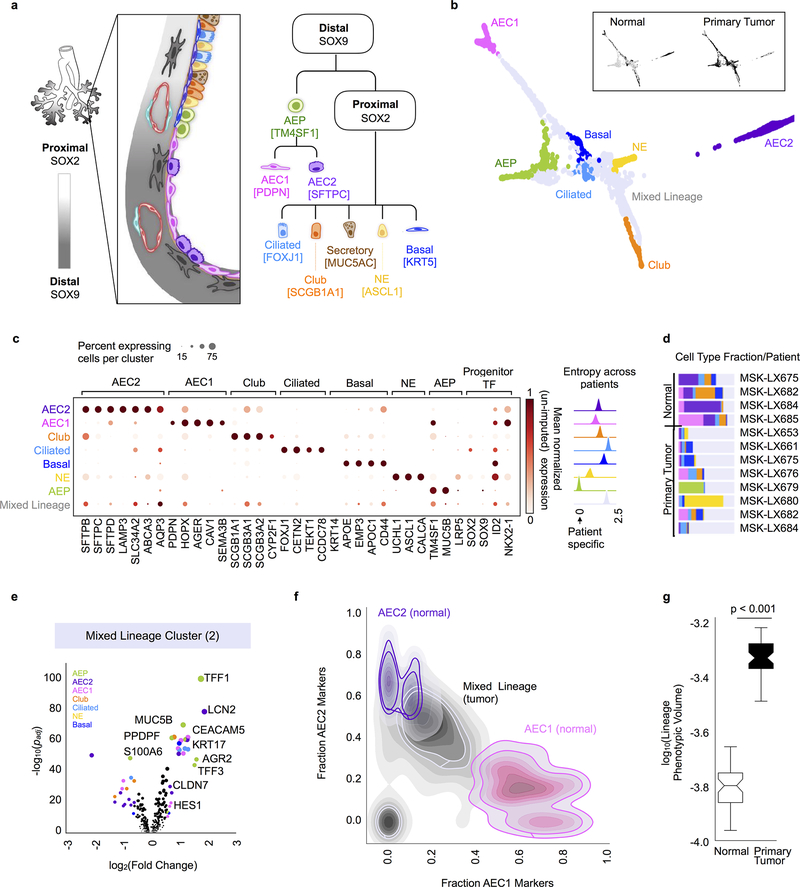
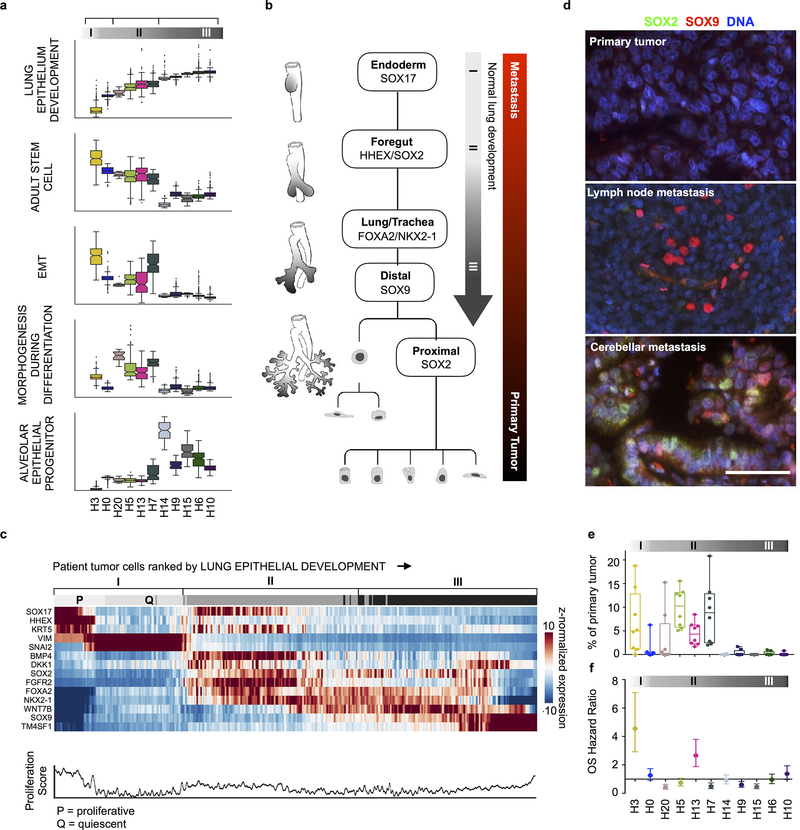
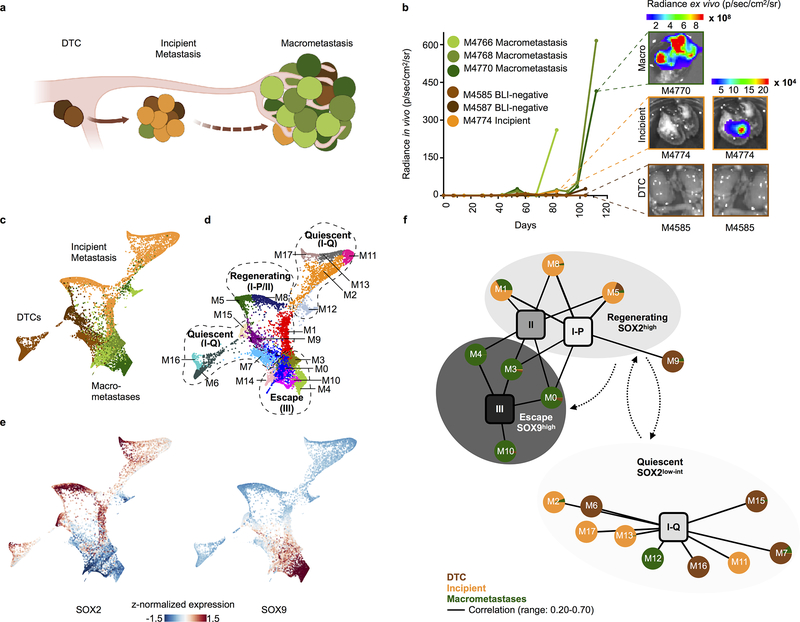
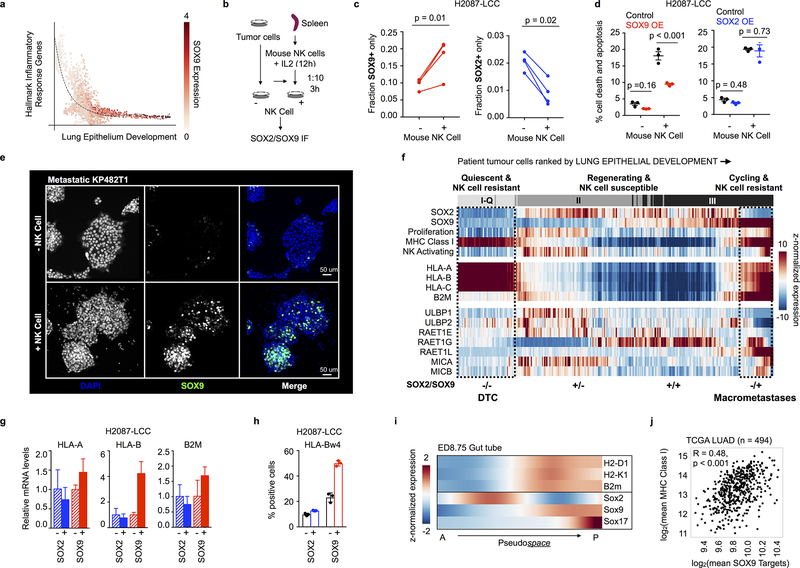
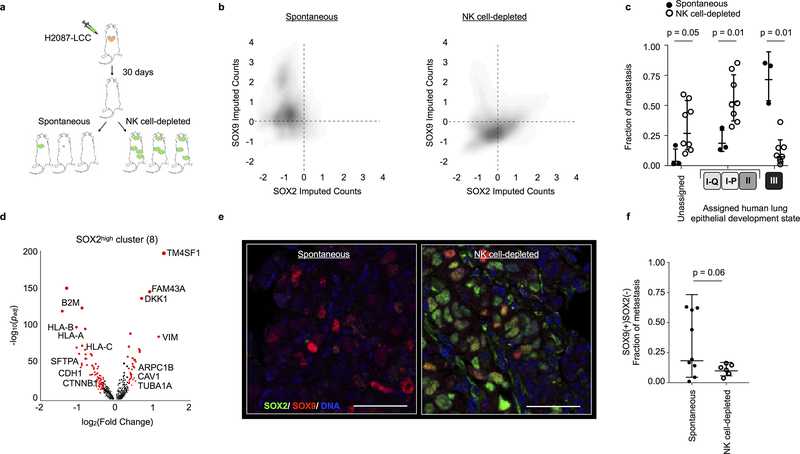
Developmental processes underlying normal tissue regeneration have been implicated in cancer, but the degree of their enactment during tumor progression and under the selective pressures of immune surveillance, remain unknown. Here we show that human primary lung adenocarcinomas are characterized by the emergence of regenerative cell types, typically seen in response to lung injury, and by striking infidelity among transcription factors specifying most alveolar and bronchial epithelial lineages. In contrast, metastases are enriched for key endoderm and lung-specifying transcription factors, SOX2 and SOX9, and recapitulate more primitive transcriptional programs spanning stem-like to regenerative pulmonary epithelial progenitor states. This developmental continuum mirrors the progressive stages of spontaneous outbreak from metastatic dormancy in a mouse model and exhibits SOX9-dependent resistance to natural killer cells. Loss of developmental stage-specific constraint in macrometastases triggered by natural killer cell depletion suggests a dynamic interplay between developmental plasticity and immune-mediated pruning during metastasis.
Conflict of interest statement
Competing Interests
J.M. is a scientific advisor and owns company stock in Scholar Rock. C.M.R. has consulted with AbbVie, Amgen, Ascentage, Astra Zeneca, BMS, Celgene, Daiichi Sankyo, Genentech/Roche, Ipsen, Loxo, and Pharmar, and is on the scientific advisory boards of Elucida and Harpoon. S.F.B. owns equity in, receives compensation from, and serves as a consultant, board member, and a scientific advisory board member for Volastra Therapeutics Inc. He also has consulted for Sanofi. All other authors declare no competing conflicts.
Figures
Comment in
-
Developmental Plasticity Mediates Lung Cancer Metastatic Potential.Cancer Discov. 2020 Apr;10(4):486. doi: 10.1158/2159-8290.CD-RW2020-028. Epub 2020 Feb 21. Cancer Discov. 2020. PMID: 32086313
References
METHODS-ONLY REFERENCES
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
