

The evolutionary plasticity of chromosome metabolism allows adaptation to constitutive DNA replication stress
- PMID: 32043971
- PMCID: PMC7069727
- DOI: 10.7554/eLife.51963
The evolutionary plasticity of chromosome metabolism allows adaptation to constitutive DNA replication stress
Abstract
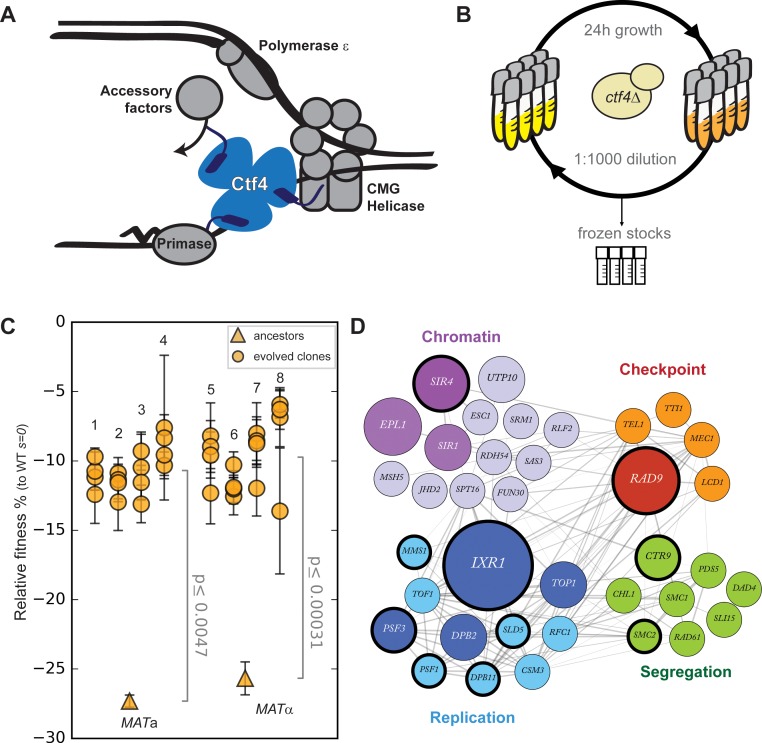
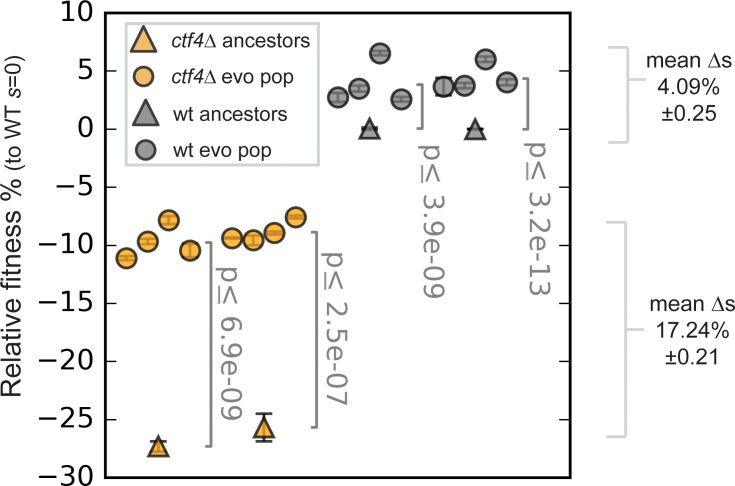
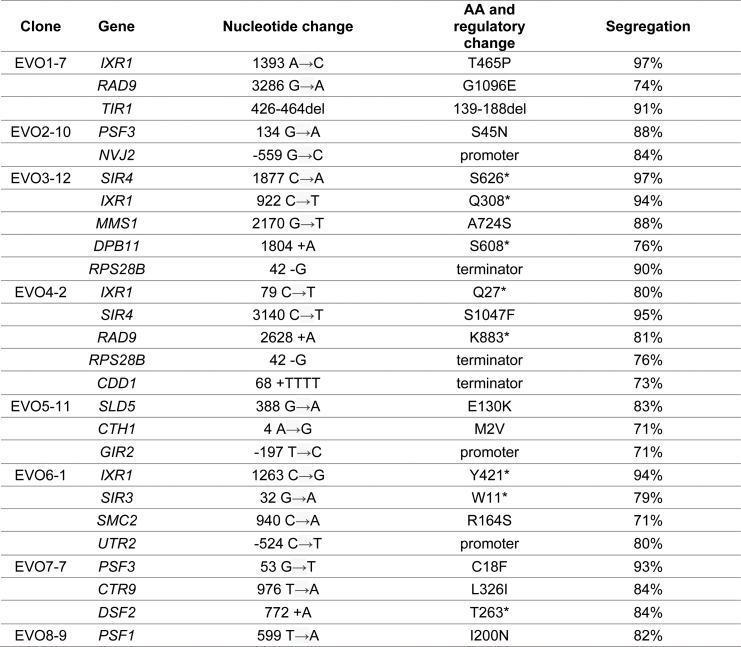
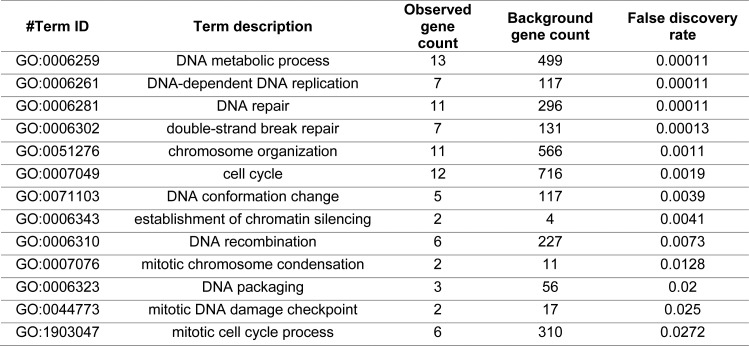
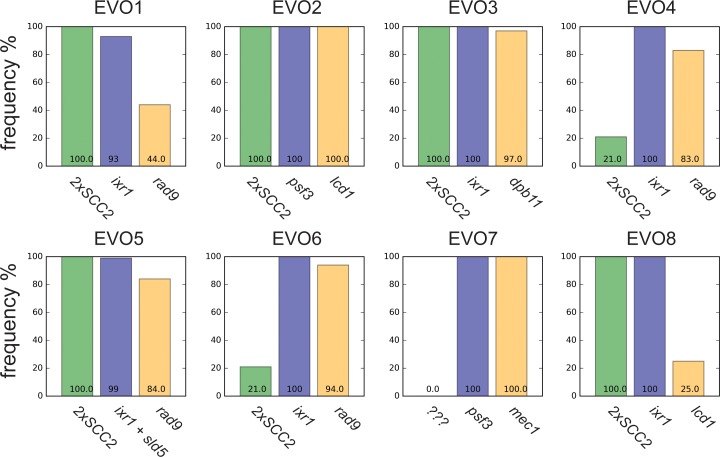
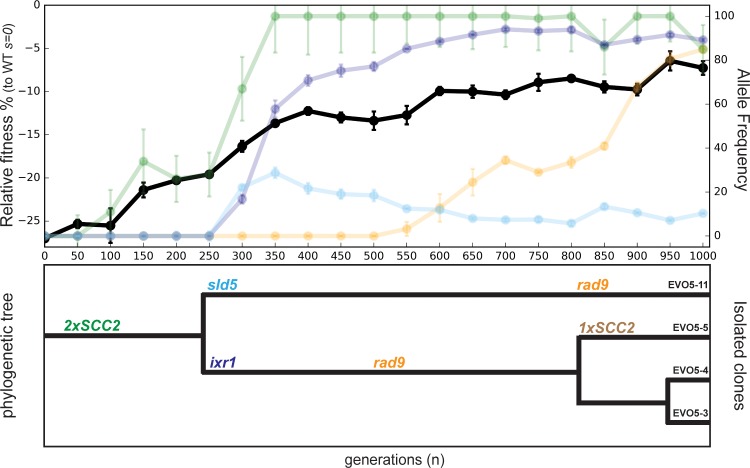
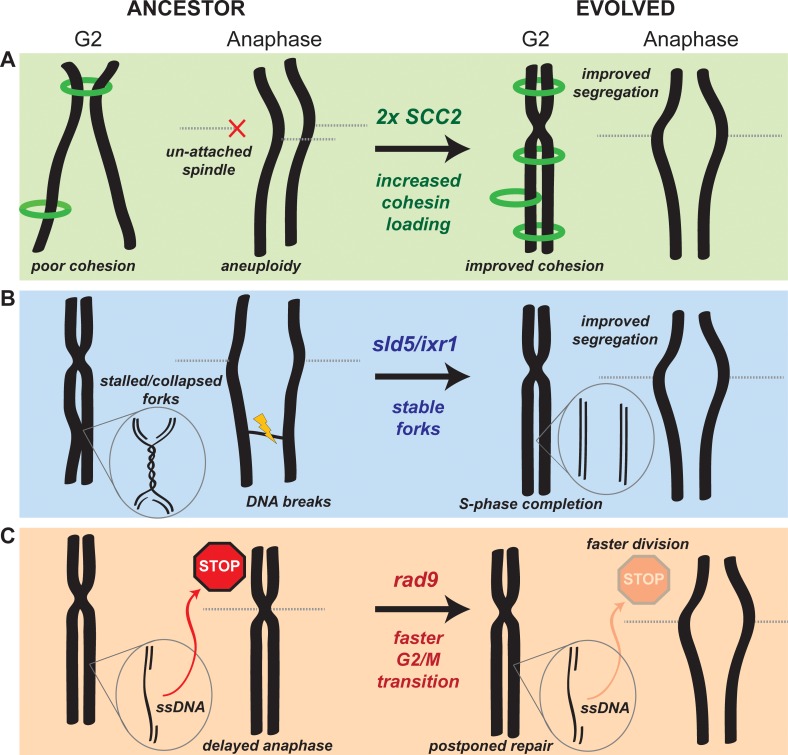
Many biological features are conserved and thus considered to be resistant to evolutionary change. While rapid genetic adaptation following the removal of conserved genes has been observed, we often lack a mechanistic understanding of how adaptation happens. We used the budding yeast, Saccharomyces cerevisiae, to investigate the evolutionary plasticity of chromosome metabolism, a network of evolutionary conserved modules. We experimentally evolved cells constitutively experiencing DNA replication stress caused by the absence of Ctf4, a protein that coordinates the enzymatic activities at replication forks. Parallel populations adapted to replication stress, over 1000 generations, by acquiring multiple, concerted mutations. These mutations altered conserved features of two chromosome metabolism modules, DNA replication and sister chromatid cohesion, and inactivated a third, the DNA damage checkpoint. The selected mutations define a functionally reproducible evolutionary trajectory. We suggest that the evolutionary plasticity of chromosome metabolism has implications for genome evolution in natural populations and cancer.
Keywords: DNA replication; S. cerevisiae; cell cycle checkpoints; chromosome segregation; evolutionary biology; evolvability; experimental evolution; genetics; genome evolution; genomics.
Plain language summary
All plants, animals and fungi share a common ancestor, and though they have evolved to become very distinct over billions of years, they all share the essential machinery needed for cells to grow and divide. At the heart of this is the complex interaction of proteins involved in DNA replication, the process of duplicating the genetic material every time a cell divides. DNA replication needs to be done with great care, with error rates as small as one mistake in a billion. Otherwise, mutations can accumulate in the genome, causing problems for long-term survival. Despite the overall principles of DNA replication remaining the same, the underlying mechanisms vary across different organisms. Given the precision and complexity of replicating DNA, it was not clear how the process had evolved mechanistic differences. Fumasoni and Murray set out to answer this by forcing a strain of budding yeast to evolve by removing the gene for an important, but not essential, component of DNA replication. The cells were still able to reproduce, but they were hampered by this mutation. Fumasoni and Murray studied the yeast after it had reproduced for a thousand generations, giving it enough time to acquire new mutations that would allow it to compensate for the initial defects. In eight separate samples, the yeast had made many of the same changes in order to overcome the original mutation. These mutations altered conserved features of DNA replication and the segregation of genetic material and inactivated a third feature that would normally protect the cell against the accumulation of damaged DNA. These findings show how reproducible the evolutionary pathways can be in a controlled, laboratory environment and that cells can evolve quickly after conserved processes in the cell are damaged. The behavior of the mutated yeast mimicked that of cancer cells, which are often struggling to adapt to mutations in their replication machinery. Studying the rapid evolution that follows genetic perturbations could help researchers to better deal with challenges in cancer treatment and the development of antibiotic resistance in bacteria, as well as leading to a deeper understanding of both evolution and cell biology.
© 2020, Fumasoni and Murray.
Conflict of interest statement
MF, AM No competing interests declared
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
