

Plasmids Shaped the Recent Emergence of the Major Nosocomial Pathogen Enterococcus faecium
- PMID: 32047136
- PMCID: PMC7018651
- DOI: 10.1128/mBio.03284-19
Plasmids Shaped the Recent Emergence of the Major Nosocomial Pathogen Enterococcus faecium
Abstract
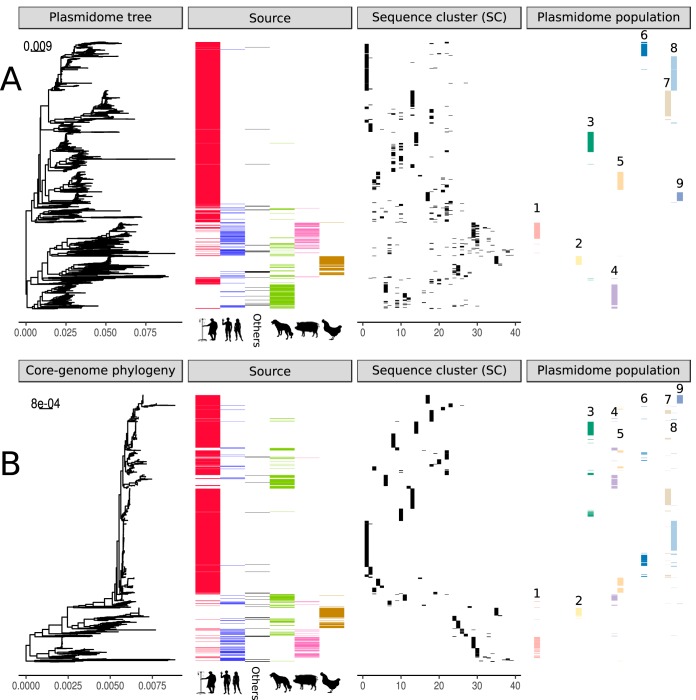
Enterococcus faecium is a gut commensal of humans and animals but is also listed on the WHO global priority list of multidrug-resistant pathogens. Many of its antibiotic resistance traits reside on plasmids and have the potential to be disseminated by horizontal gene transfer. Here, we present the first comprehensive population-wide analysis of the pan-plasmidome of a clinically important bacterium, by whole-genome sequence analysis of 1,644 isolates from hospital, commensal, and animal sources of E. faecium Long-read sequencing on a selection of isolates resulted in the completion of 305 plasmids that exhibited high levels of sequence modularity. We further investigated the entirety of all plasmids of each isolate (plasmidome) using a combination of short-read sequencing and machine-learning classifiers. Clustering of the plasmid sequences unraveled different E. faecium populations with a clear association with hospitalized patient isolates, suggesting different optimal configurations of plasmids in the hospital environment. The characterization of these populations allowed us to identify common mechanisms of plasmid stabilization such as toxin-antitoxin systems and genes exclusively present in particular plasmidome populations exemplified by copper resistance, phosphotransferase systems, or bacteriocin genes potentially involved in niche adaptation. Based on the distribution of k-mer distances between isolates, we concluded that plasmidomes rather than chromosomes are most informative for source specificity of E. faeciumIMPORTANCEEnterococcus faecium is one of the most frequent nosocomial pathogens of hospital-acquired infections. E. faecium has gained resistance against most commonly available antibiotics, most notably, against ampicillin, gentamicin, and vancomycin, which renders infections difficult to treat. Many antibiotic resistance traits, in particular, vancomycin resistance, can be encoded in autonomous and extrachromosomal elements called plasmids. These sequences can be disseminated to other isolates by horizontal gene transfer and confer novel mechanisms to source specificity. In our study, we elucidated the total plasmid content, referred to as the plasmidome, of 1,644 E. faecium isolates by using short- and long-read whole-genome technologies with the combination of a machine-learning classifier. This was fundamental to investigate the full collection of plasmid sequences present in our collection (pan-plasmidome) and to observe the potential transfer of plasmid sequences between E. faecium hosts. We observed that E. faecium isolates from hospitalized patients carried a larger number of plasmid sequences compared to that from other sources, and they elucidated different configurations of plasmidome populations in the hospital environment. We assessed the contribution of different genomic components and observed that plasmid sequences have the highest contribution to source specificity. Our study suggests that E. faecium plasmids are regulated by complex ecological constraints rather than physical interaction between hosts.
Keywords: Enterococcus faecium; long-read sequencing; machine learning; nosocomial pathogen; plasmidome; source specificity.
Copyright © 2020 Arredondo-Alonso et al.
Figures




References
-
- Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, Edwards JR, Sievert DM. 2016. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014. Infect Control Hosp Epidemiol 37:1288–1301. doi: 10.1017/ice.2016.174. - DOI - PMC - PubMed
-
- Palmer KL, Godfrey P, Griggs A, Kos VN, Zucker J, Desjardins C, Cerqueira G, Gevers D, Walker S, Wortman J, Feldgarden M, Haas B, Birren B, Gilmore MS. 2012. Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. mBio 3:e00318-11. doi: 10.1128/mBio.00318-11. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
