

doi: 10.1107/S1600576719014092.
Mercury 4.0: from visualization to analysis, design and prediction
Affiliations
- PMID: 32047413
- PMCID: PMC6998782
- DOI: 10.1107/S1600576719014092
Item in Clipboard
Mercury 4.0: from visualization to analysis, design and prediction
J Appl Crystallogr.
.
Abstract
The program Mercury, developed at the Cambridge Crystallographic Data Centre, was originally designed primarily as a crystal structure visualization tool. Over the years the fields and scientific communities of chemical crystallography and crystal engineering have developed to require more advanced structural analysis software. Mercury has evolved alongside these scientific communities and is now a powerful analysis, design and prediction platform which goes a lot further than simple structure visualization.
Keywords: Mercury; computer programs; crystal packing; crystal structure visualization; structure comparison.
© Clare F. Macrae et al. 2020.
Figures
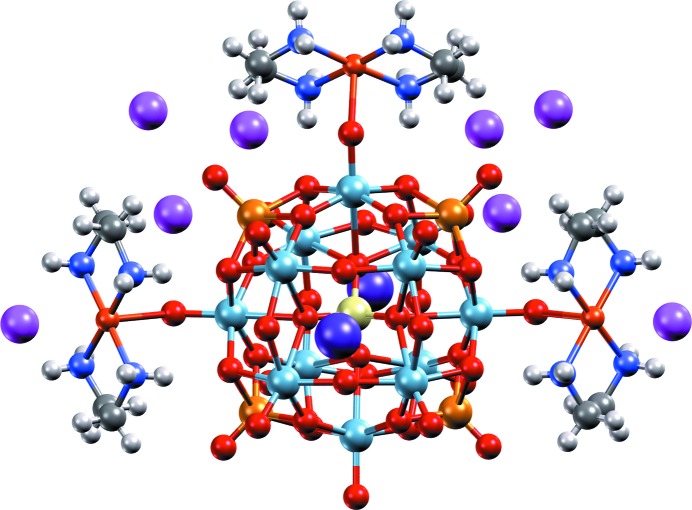
High-resolution image, generated with Mercury and POV-Ray, of a heteropolyoxoniobate-based system (CSD refcode LOFHOF; Zhang et al., 2014 ▸) using the ‘Shiny’ material property.
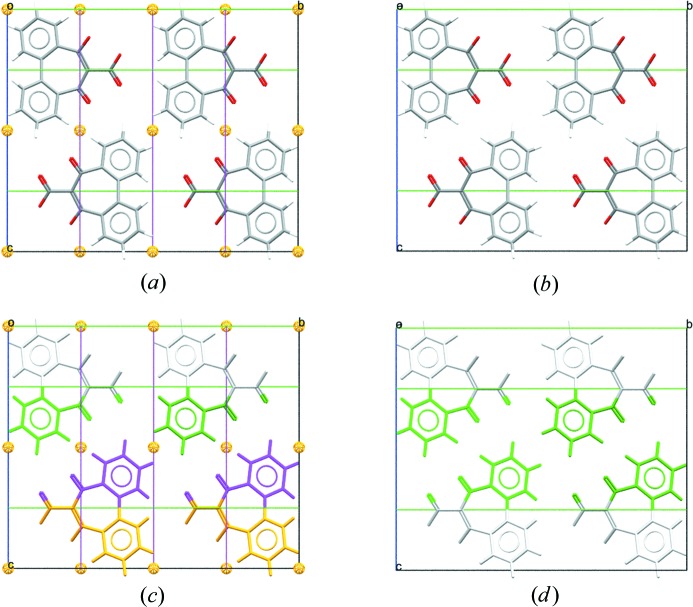
Packing diagrams for CSD refcode SUGCEC (Mochida et al., 1992 ▸). (a) Unedited, presented in the original space group of C2/c with symmetry elements displayed. Note that the molecule lies on a twofold axis parallel to the b axis. (b) Unedited, with molecules coloured by symmetry operation (other than centring) and symmetry elements shown. (c) Edited to the subgroup C2, setting 1, origin choice [0, 0, 1/4], with symmetry elements displayed. (d) Edited to the subgroup C2, with molecules coloured by symmetry operation (other than centring). Transforming to C2 from space group C2/c retains only the twofold axes of the space group and increases the number of formula units in the asymmetric unit to two.
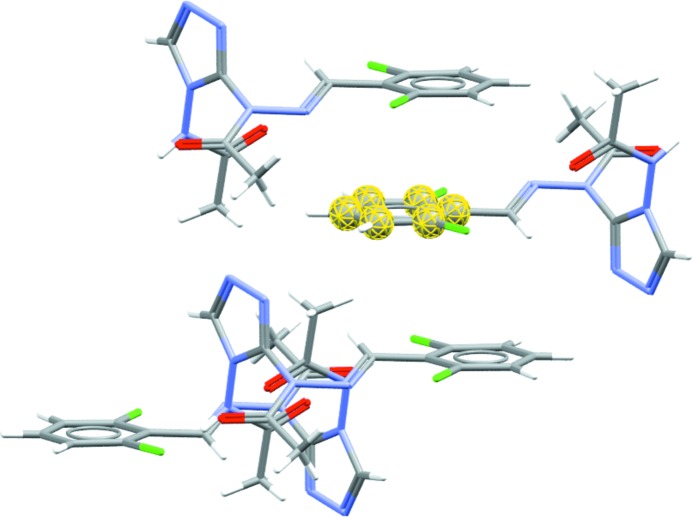
Diagram showing the molecular shell, calculated using a radius of van der Waals + 0.5 Å, from the phenyl ring fragment of a molecule in AABHTZ (Werner, 1976 ▸) (highlighted in yellow). The packing shell generated here highlights the aromatic interactions present in the structure. Full packing shells of molecules can be calculated by selecting a molecule (rather than a fragment) as the base unit.
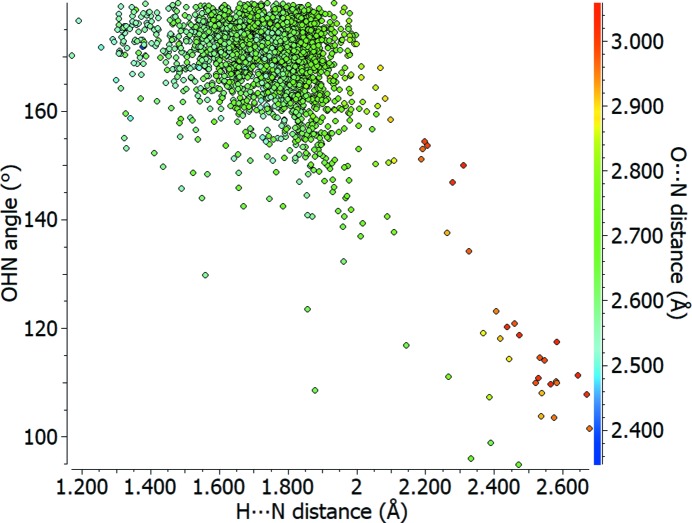
Scatterplot of O—H⋯N angle (°) against H⋯N distance (Å) for carboxylic acid to pyridine type nitrogen–hydrogen bonds (O=C—OH⋯N=C), with the O⋯N distance (Å) shown using a colour scale.
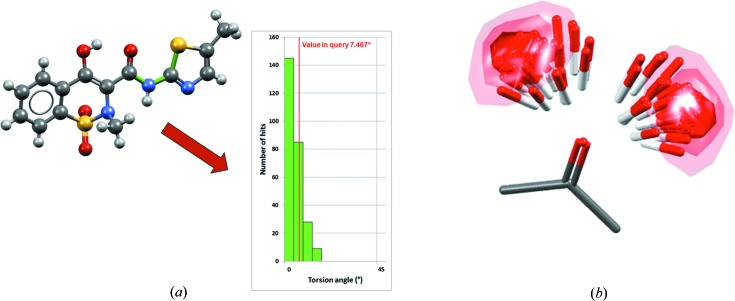
Illustration of CSD knowledge bases, showing (a) the Mogul distribution relating to a specific S—C—N—C torsion angle (bonds in the torsion are coloured green) in the meloxicam succinic acid co-crystal (CSD refcode ENICOU; Cheney et al., 2010 ▸) and (b) the IsoStar scatterplot relating to a ketone central group interacting with an alcohol probe group (contact density shown as contour surfaces).
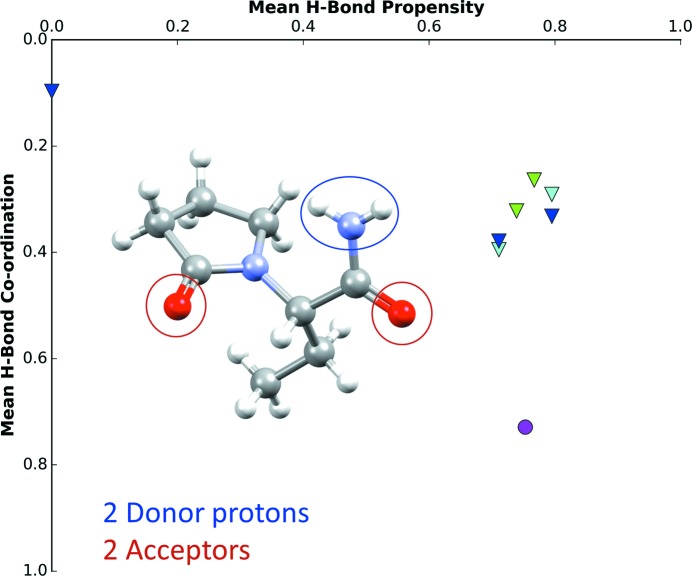
Hydrogen-bond propensity chart of levetiracetam (CSD refcode OMIVUB; Song et al., 2003 ▸). The magenta circle represents the observed structure.
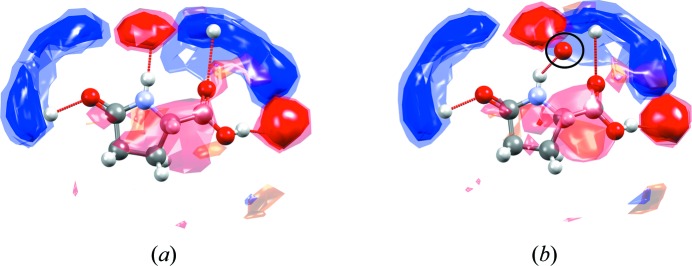
Full interaction maps shown around one of the molecules of l -pyroglutamic acid in (a) the (stable) α form (CSD refcode LPYGLU07, Z′ = 3) and (b) the (metastable) β form of the compound (CSD refcode LPYGLU08, Z′ = 1) (Panda et al., 2015 ▸). The circled oxygen acceptor in the interaction map diagram shown for the β form (b) is observed outside of any preferred acceptor region of the maps (red contours), indicating that this hydrogen-bonding interaction has a sub-optimal geometry.
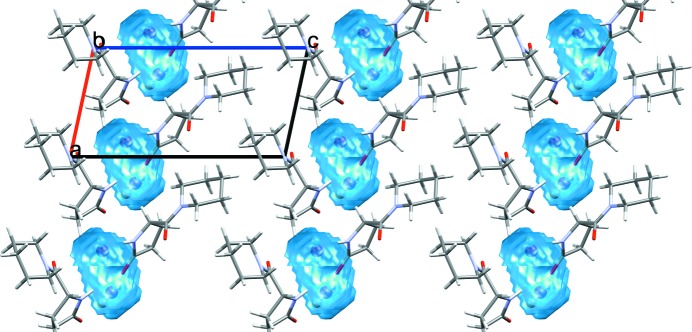
Calculated water space for fasoracetam monohydrate (CSD refcode PAPNIG; Harmsen et al., 2017 ▸) with water molecules occupying discrete pockets within the crystal structure.

Calculated solvent space for bis[bis(4-chlorobenzenesulfonyl)amine] dimethylsulfoxide (space shown in cyan) nitromethane (space shown in red) solvate (CSD refcode XUKZIM; Hamann et al., 2002 ▸).
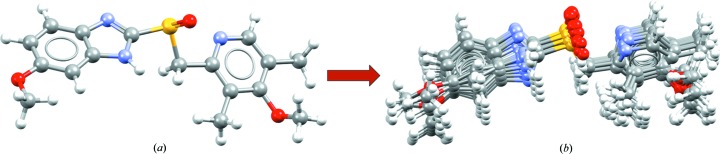
Conformer generation for the molecule omeprazole, starting from (a) the molecular conformation in a known crystal structure (CSD refocde VAYXOI; Ohishi et al., 1989 ▸) and generating (b) a diverse conformer ensemble including the 15 highest ranked conformers.
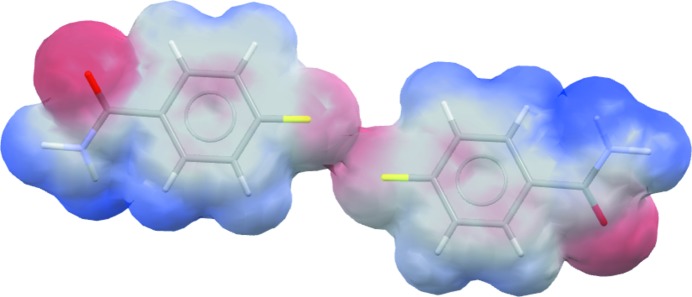
The electrostatic potential mapped onto the van der Waals surface of 4-fluorobenzamide (CSD refcode BENAFP; Takaki et al., 1965 ▸), illustrating the repulsive nature of the F⋯F contact (centre).
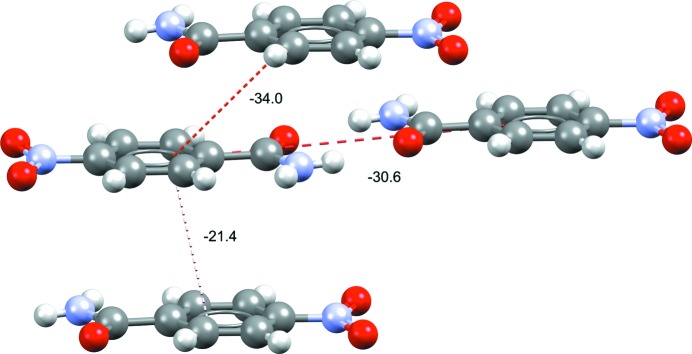
Dimer intermolecular interaction energies (kJ mol−1) of the top three interaction energies in form I of 4-nitrobenzamide (refcode NTBZAM10; Di Rienzo et al., 1977 ▸). Stacking-related dimers are seen to be close in interaction energy to the observed hydrogen-bonded dimers.
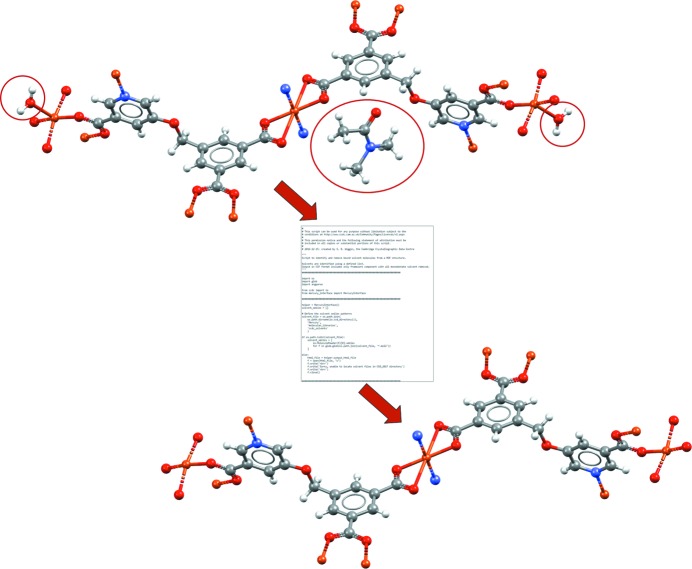
A CSD Python API script that can be run within Mercury. This example shows a script allowing automated removal of both unbound and bound solvent molecules (matching to a specified solvent library) from a MOF structure. The example shown is of a copper MOF structure (CSD refcode BEXSII; Patra et al., 2003 ▸). In this case the Python script takes the unedited crystal structure as input (top), removes the unbound dimethylformamide solvent (circled) as well as the two singly bound water molecules (circled) and outputs the edited crystal structure for viewing in Mercury (bottom).
References
-
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. - PubMed
-
- Allen, F. H., Johnson, O., Shields, G. P., Smith, B. R. & Towler, M. (2004). J. Appl. Cryst. 37, 335–338.
-
- Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002). Acta Cryst. B58, 389–397. - PubMed
-
- Bruno, I. J., Cole, J. C., Kessler, M., Luo, J., Motherwell, W. D. S., Purkis, L. H., Smith, B. R., Taylor, R., Cooper, R. I., Harris, S. E. & Orpen, A. G. (2004). J. Chem. Inf. Comput. Sci. 44, 2133–2144. - PubMed
-
- Bruno, I. J., Cole, J. C., Lommerse, J. P. M., Rowland, R. S., Taylor, R. & Verdonk, M. L. (1997). J. Comput. Aided Mol. Des. 11, 525–537. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources