

Multiple Myeloma: Available Therapies and Causes of Drug Resistance
- PMID: 32050631
- PMCID: PMC7072128
- DOI: 10.3390/cancers12020407
Multiple Myeloma: Available Therapies and Causes of Drug Resistance
Abstract
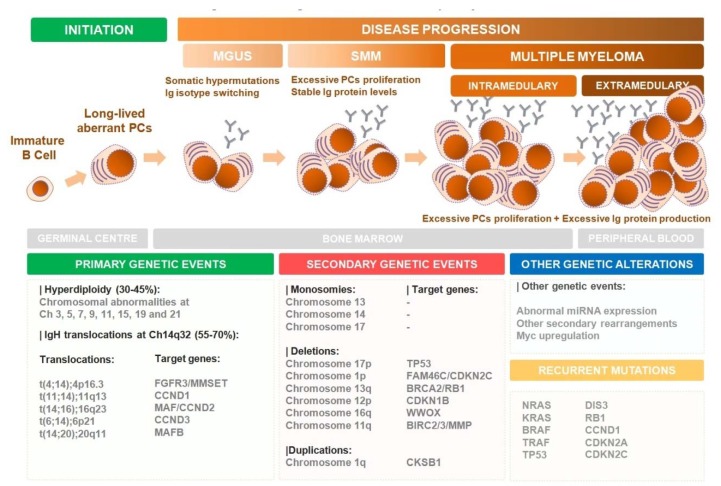
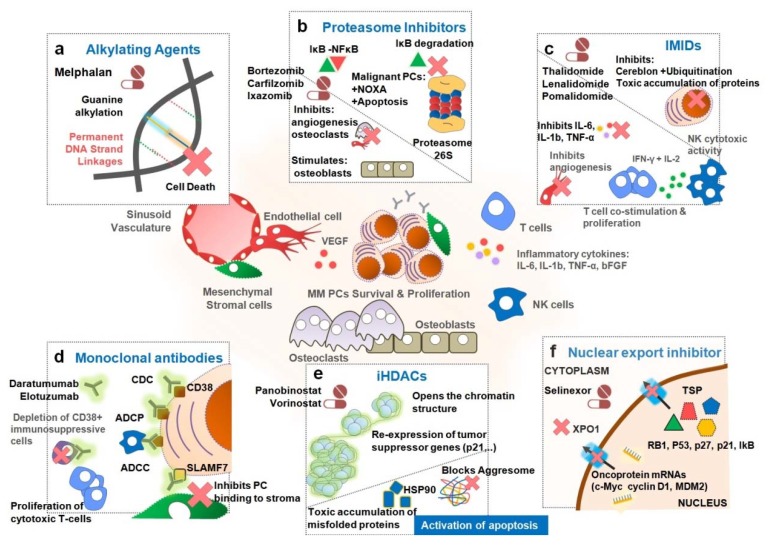
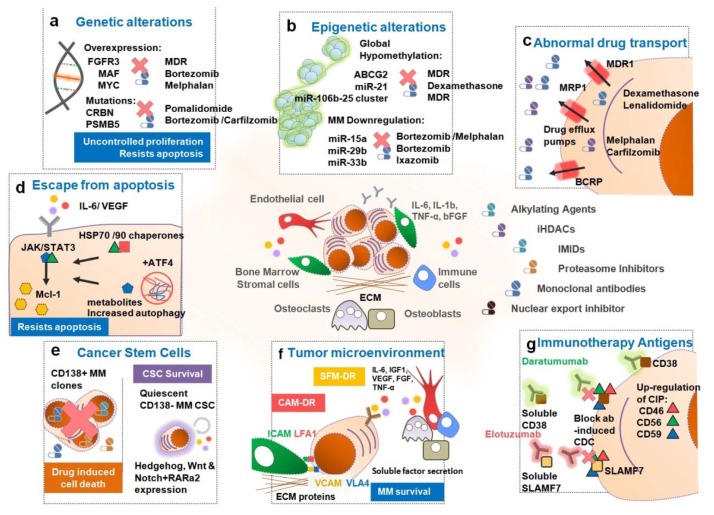
Multiple myeloma (MM) is the second most common blood cancer. Treatments for MM include corticosteroids, alkylating agents, anthracyclines, proteasome inhibitors, immunomodulatory drugs, histone deacetylase inhibitors and monoclonal antibodies. Survival outcomes have improved substantially due to the introduction of many of these drugs allied with their rational use. Nonetheless, MM patients successively relapse after one or more treatment regimens or become refractory, mostly due to drug resistance. This review focuses on the main drugs used in MM treatment and on causes of drug resistance, including cytogenetic, genetic and epigenetic alterations, abnormal drug transport and metabolism, dysregulation of apoptosis, autophagy activation and other intracellular signaling pathways, the presence of cancer stem cells, and the tumor microenvironment. Furthermore, we highlight the areas that need to be further clarified in an attempt to identify novel therapeutic targets to counteract drug resistance in MM patients.
Keywords: drug resistance; drug response; multiple myeloma; treatment.
Conflict of interest statement
R.B.: Grants and research fund: Celgene, AMGEN/SPH/APCL; Advisory boards—AMGEN, Celgene, Janssen and Takeda; Speaker honoraria—AMGEN, Celgene, Janssen and Takeda. J.E.G.: Speaker’s bureau—Abbvie, Janssen, Pfizer, Roche; Advisory boards—Abbvie, Pfizer, Roche. M.H.V., R.B. and H.C. are members of the research team of a project financed by Celgene and M.H.V. is member of the team of a grant co-financed by AMGEN. These companies had no role in the decision to publish nor were they involved in the writing of this manuscript. The authors declare no conflict of interest.
Figures
References
-
- Kumar S., Paiva B., Anderson K.C., Durie B., Landgren O., Moreau P., Munshi N., Lonial S., Bladé J., Mateos M.-V., et al. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016;17:e328–e346. doi: 10.1016/S1470-2045(16)30206-6. - DOI - PubMed
-
- Landgren O., Graubard B., Katzmann J., Kyle R., Ahmadizadeh I., Clark R., Kumar S., Dispenzieri A., Greenberg A., Therneau T., et al. Racial disparities in the prevalence of monoclonal gammopathies: A population-based study of 12 482 persons from the National Health and Nutritional Examination Survey. Leukemia. 2014;28:1537–1542. doi: 10.1038/leu.2014.34. - DOI - PMC - PubMed
-
- Kumar S., Dispenzieri A., Lacy M., Gertz M., Buadi F., Pandey S., Kapoor P., Dingli D., Hayman S., Leung N., et al. Continued improvement in survival in multiple myeloma: Changes in early mortality and outcomes in older patients. Leukemia. 2014;28:1122–1128. doi: 10.1038/leu.2013.313. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
