

Vitamin D receptor targets hepatocyte nuclear factor 4α and mediates protective effects of vitamin D in nonalcoholic fatty liver disease
- PMID: 32051143
- PMCID: PMC7086018
- DOI: 10.1074/jbc.RA119.011487
Vitamin D receptor targets hepatocyte nuclear factor 4α and mediates protective effects of vitamin D in nonalcoholic fatty liver disease
Abstract
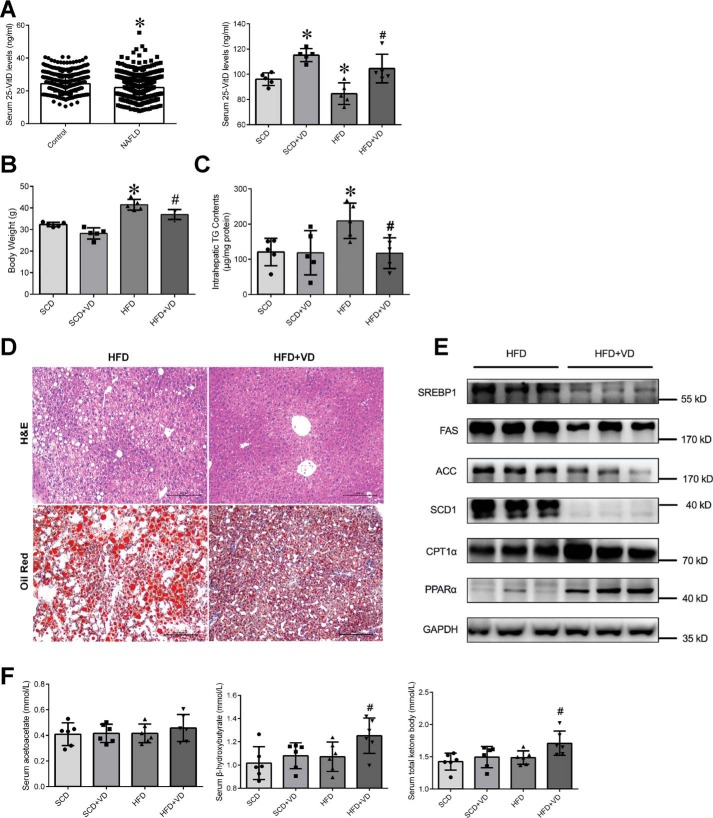
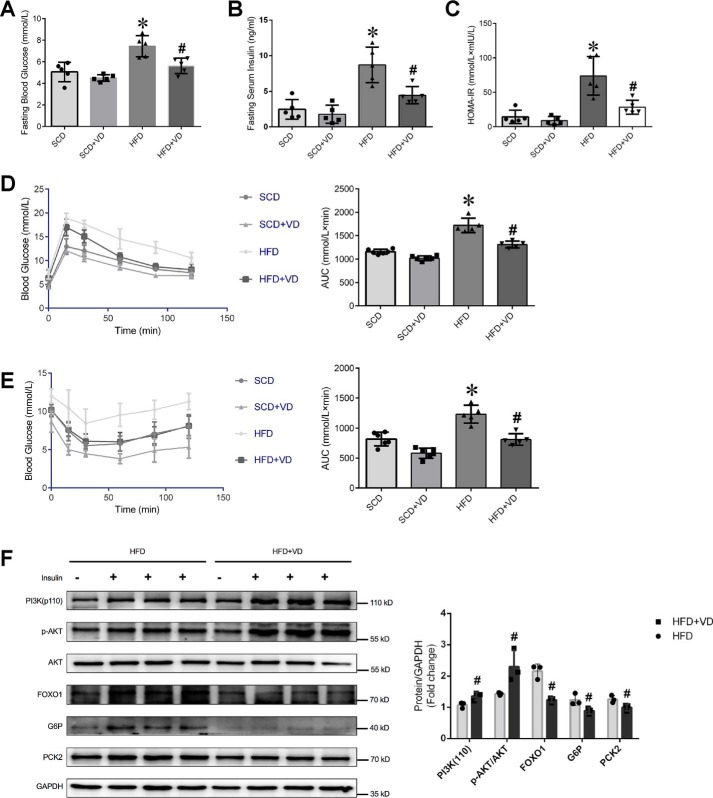
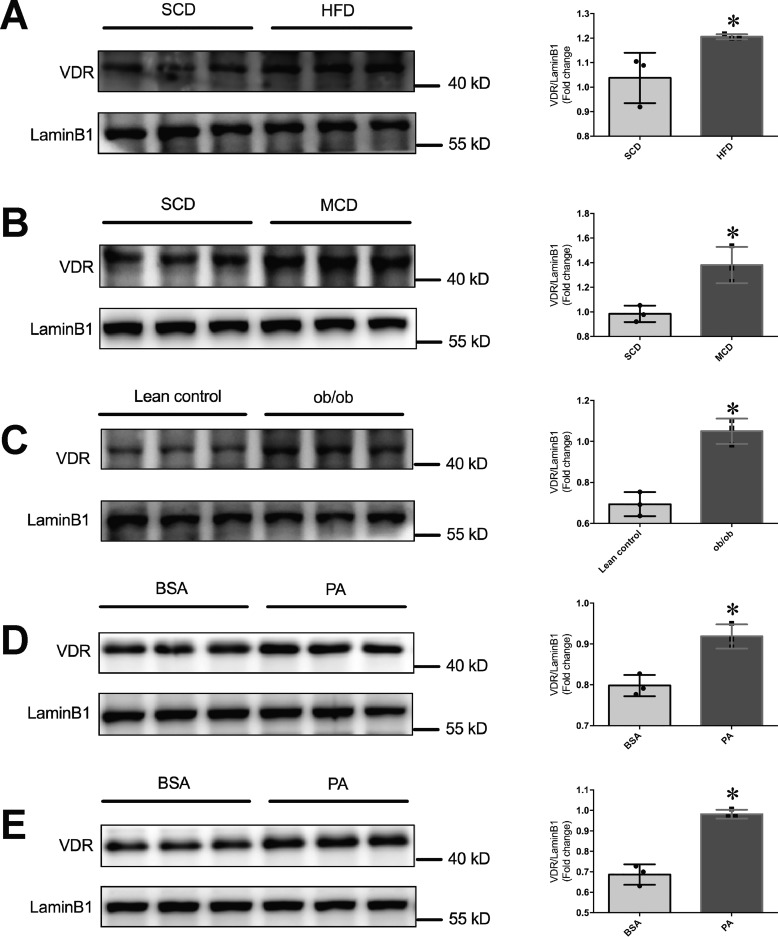
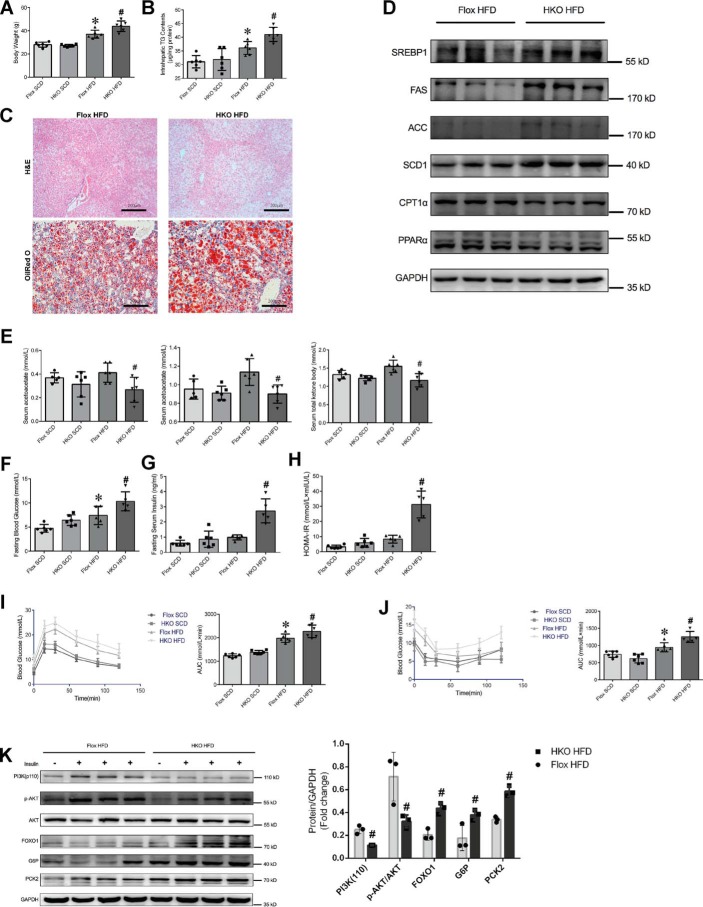
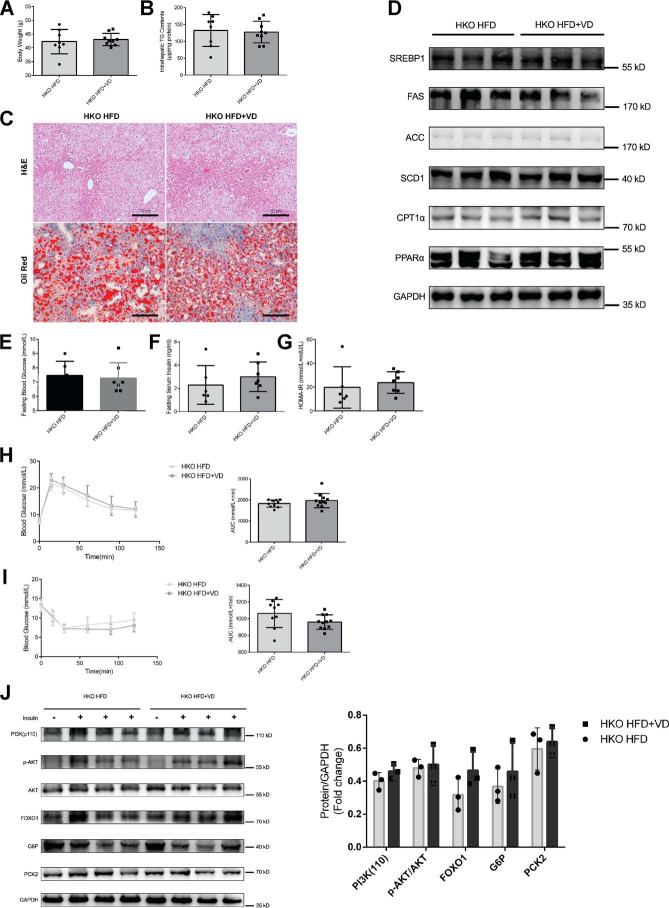
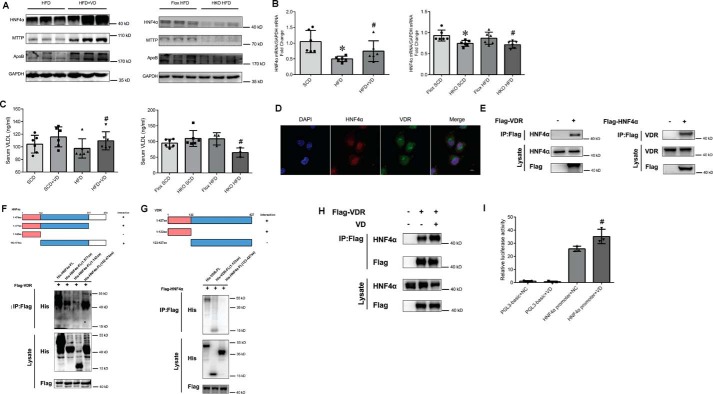
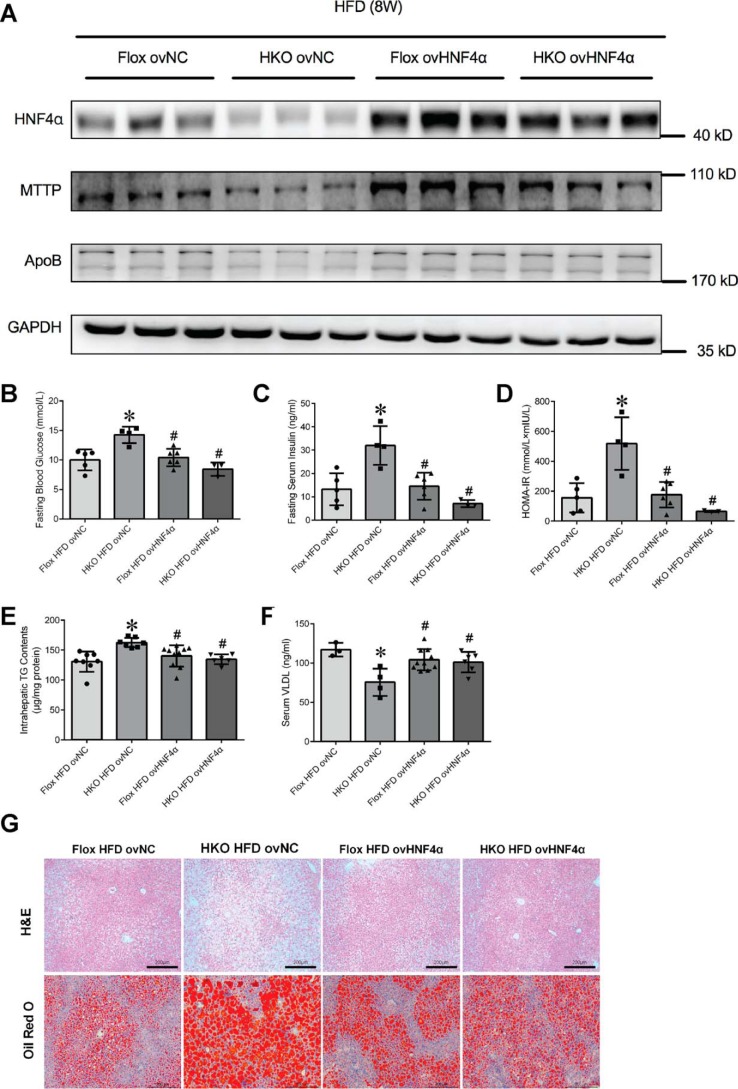
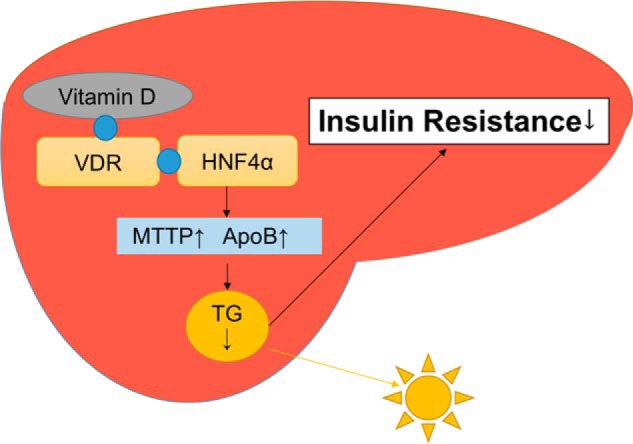
Epidemiological studies have suggested a link between vitamin D deficiency and increased risk for nonalcoholic fatty liver disease (NAFLD); however, the underlying mechanisms have remained unclear. Here, using both clinical samples and experimental rodent models along with several biochemical approaches, we explored the specific effects and mechanisms of vitamin D deficiency in NAFLD pathology. Serum vitamin D levels were significantly lower in individuals with NAFLD and in high-fat diet (HFD)-fed mice than in healthy controls and chow-fed mice, respectively. Vitamin D supplementation ameliorated HFD-induced hepatic steatosis and insulin resistance in mice. Hepatic expression of vitamin D receptor (VDR) was up-regulated in three models of NAFLD, including HFD-fed mice, methionine/choline-deficient diet (MCD)-fed mice, and genetically obese (ob/ob) mice. Liver-specific VDR deletion significantly exacerbated HFD- or MCD-induced hepatic steatosis and insulin resistance and also diminished the protective effect of vitamin D supplementation on NAFLD. Mechanistic experiments revealed that VDR interacted with hepatocyte nuclear factor 4 α (HNF4α) and that overexpression of HNF4α improved HFD-induced NAFLD and metabolic abnormalities in liver-specific VDR-knockout mice. These results suggest that vitamin D ameliorates NAFLD and metabolic abnormalities by activating hepatic VDR, leading to its interaction with HNF4α. Our findings highlight a potential value of using vitamin D for preventing and managing NAFLD by targeting VDR.
Keywords: gene regulation; hepatic steatosis; hepatocyte nuclear factor 4 α (HNF4α); insulin resistance; lipid metabolism; liver metabolism; metabolic syndrome; nonalcoholic fatty liver disease (NAFLD); nuclear receptor; vitamin D.
© 2020 Zhang et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Lee M. S., Han H. J., Han S. Y., Kim I. Y., Chae S., Lee C. S., Kim S. E., Yoon S. G., Park J. W., Kim J. H., Shin S., Jeong M., Ko A., Lee H. Y., Oh K. J., et al. (2018) Loss of the E3 ubiquitin ligase MKRN1 represses diet-induced metabolic syndrome through AMPK activation. Nat. Commun. 9, 3404 10.1038/s41467-018-05721-4 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
