

A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients
- PMID: 32051234
- PMCID: PMC7124470
- DOI: 10.1074/mcp.TIR119.001906
A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients
Abstract
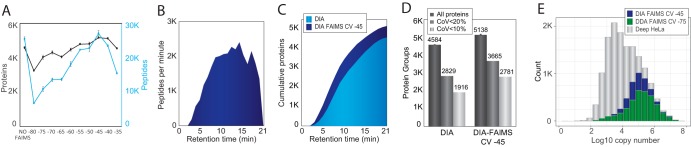
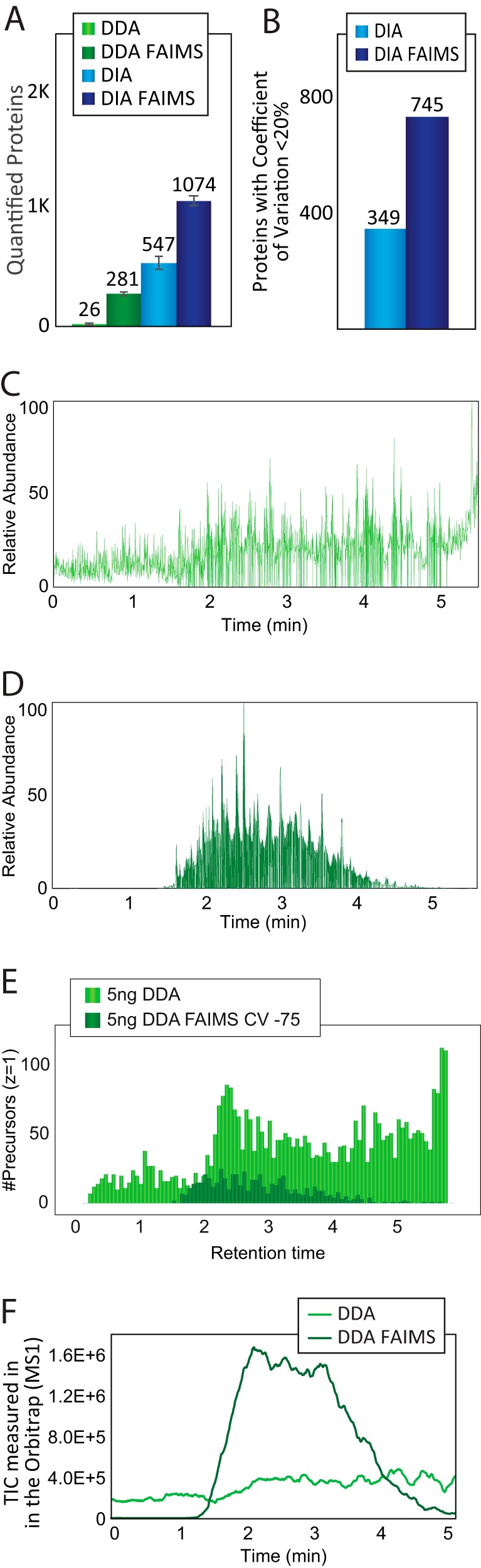
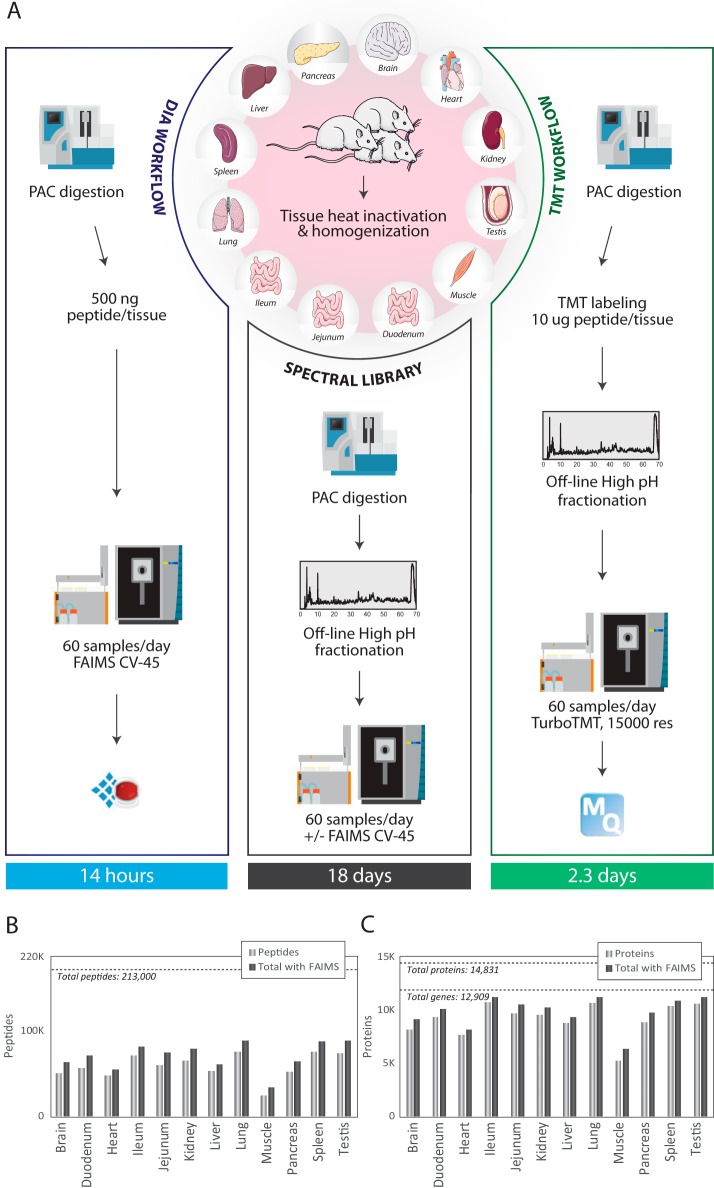
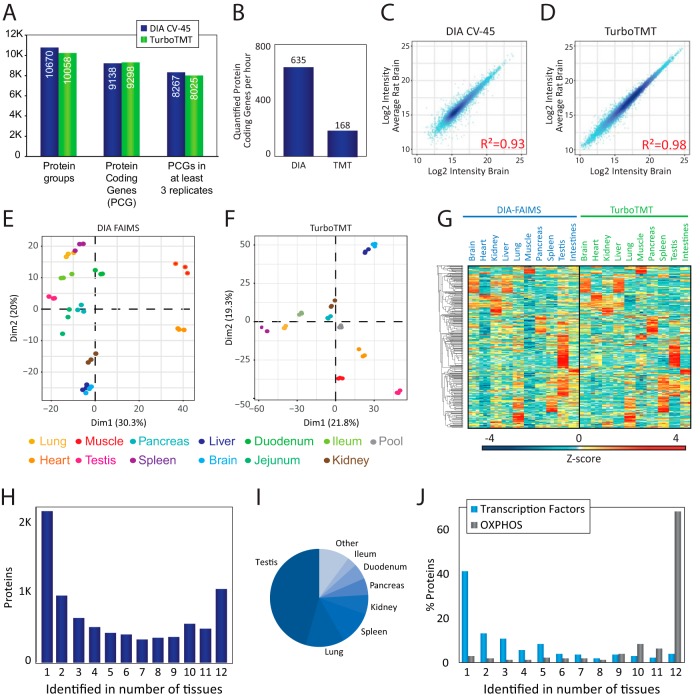
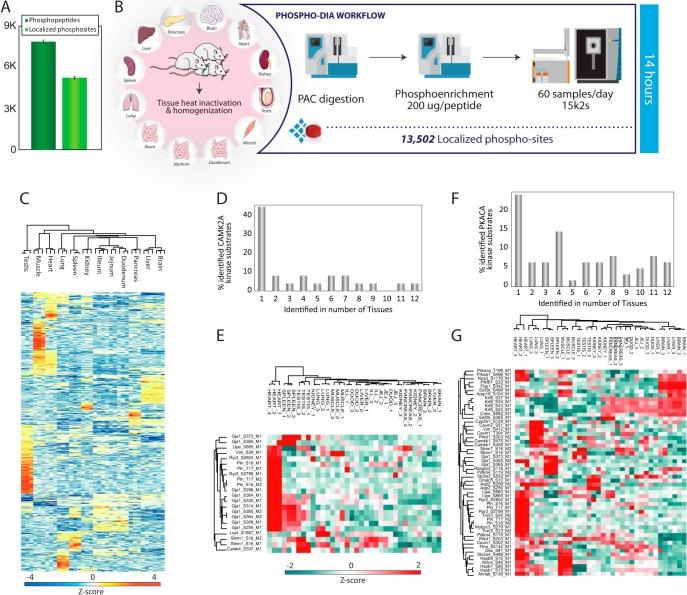
State-of-the-art proteomics-grade mass spectrometers can measure peptide precursors and their fragments with ppm mass accuracy at sequencing speeds of tens of peptides per second with attomolar sensitivity. Here we describe a compact and robust quadrupole-orbitrap mass spectrometer equipped with a front-end High Field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS) Interface. The performance of the Orbitrap Exploris 480 mass spectrometer is evaluated in data-dependent acquisition (DDA) and data-independent acquisition (DIA) modes in combination with FAIMS. We demonstrate that different compensation voltages (CVs) for FAIMS are optimal for DDA and DIA, respectively. Combining DIA with FAIMS using single CVs, the instrument surpasses 2500 peptides identified per minute. This enables quantification of >5000 proteins with short online LC gradients delivered by the Evosep One LC system allowing acquisition of 60 samples per day. The raw sensitivity of the instrument is evaluated by analyzing 5 ng of a HeLa digest from which >1000 proteins were reproducibly identified with 5 min LC gradients using DIA-FAIMS. To demonstrate the versatility of the instrument, we recorded an organ-wide map of proteome expression across 12 rat tissues quantified by tandem mass tags and label-free quantification using DIA with FAIMS to a depth of >10,000 proteins.
Keywords: DDA; DIA; FAIMS; Tandem mass spectrometry; clinical proteomics; orbitrap; pathway analysis; phosphoproteome; protein identification; proteomics; transcription.
© 2020 Bekker-Jensen et al.
Figures

References
-
- Kim M. S., Pinto S. M., Getnet D., Nirujogi R. S., Manda S. S., Chaerkady R., Madugundu A. K., Kelkar D. S., Isserlin R., Jain S., Thomas J. K., Muthusamy B., Leal-Rojas P., Kumar P., Sahasrabuddhe N. A., Balakrishnan L., Advani J., George B., Renuse S., Selvan L. D., Patil A. H., Nanjappa V., Radhakrishnan A., Prasad S., Subbannayya T., Raju R., Kumar M., Sreenivasamurthy S. K., Marimuthu A., Sathe G. J., Chavan S., Datta K. K., Subbannayya Y., Sahu A., Yelamanchi S. D., Jayaram S., Rajagopalan P., Sharma J., Murthy K. R., Syed N., Goel R., Khan A. A., Ahmad S., Dey G., Mudgal K., Chatterjee A., Huang T. C., Zhong J., Wu X., Shaw P. G., Freed D., Zahari M. S., Mukherjee K. K., Shankar S., Mahadevan A., Lam H., Mitchell C. J., Shankar S. K., Satishchandra P., Schroeder J. T., Sirdeshmukh R., Maitra A., Leach S. D., Drake C. G., Halushka M. K., Prasad T. S., Hruban R. H., Kerr C. L., Bader G. D., Iacobuzio-Donahue C. A., Gowda H., and Pandey A. (2014) A Draft Map of the human proteome. Nature 509, 575–581 - PMC - PubMed
-
- Wilhelm M., Schlegl J., Hahne H., Gholami A. M., Lieberenz M., Savitski M. M., Ziegler E., Butzmann L., Gessulat S., Marx H., Mathieson T., Lemeer S., Schnatbaum K., Reimer U., Wenschuh H., Mollenhauer M., Slotta-Huspenina J., Boese J. H., Bantscheff M., Gerstmair A., Faerber F., and Kuster B. (2014) Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587 - PubMed
-
- Kelstrup C. D., Young C., Lavallee R., Nielsen M. L., Olsen J. V. (2012) Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. J. Proteome Res. 11, 3487–3497 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
