

Variant antigen diversity in Trypanosoma vivax is not driven by recombination
- PMID: 32051413
- PMCID: PMC7015903
- DOI: 10.1038/s41467-020-14575-8
Variant antigen diversity in Trypanosoma vivax is not driven by recombination
Abstract
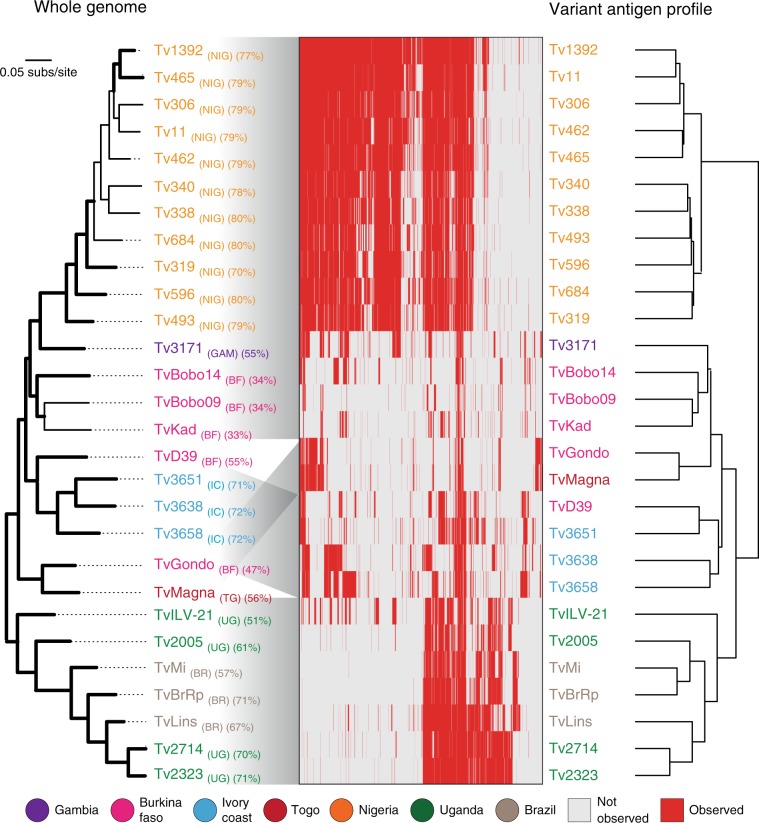
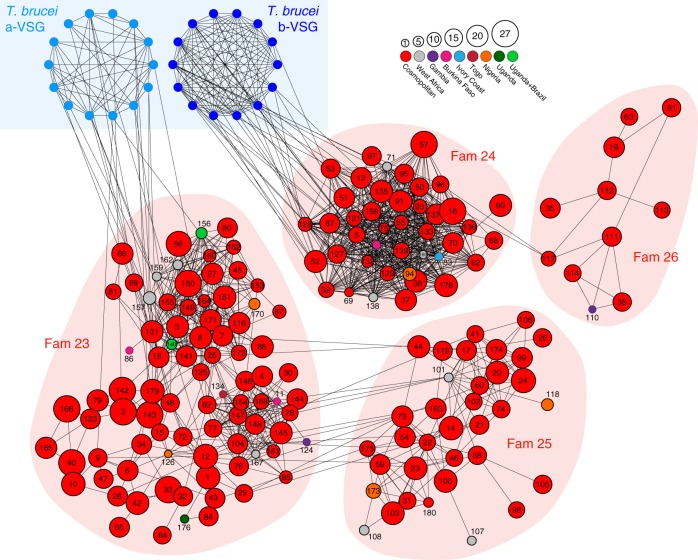
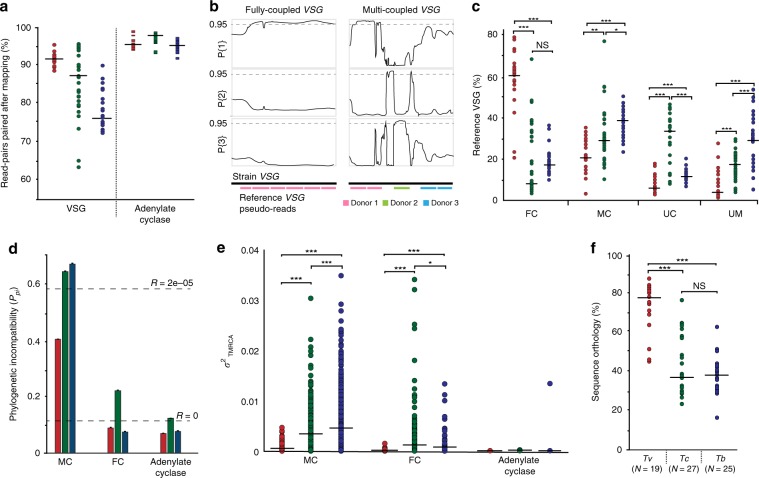
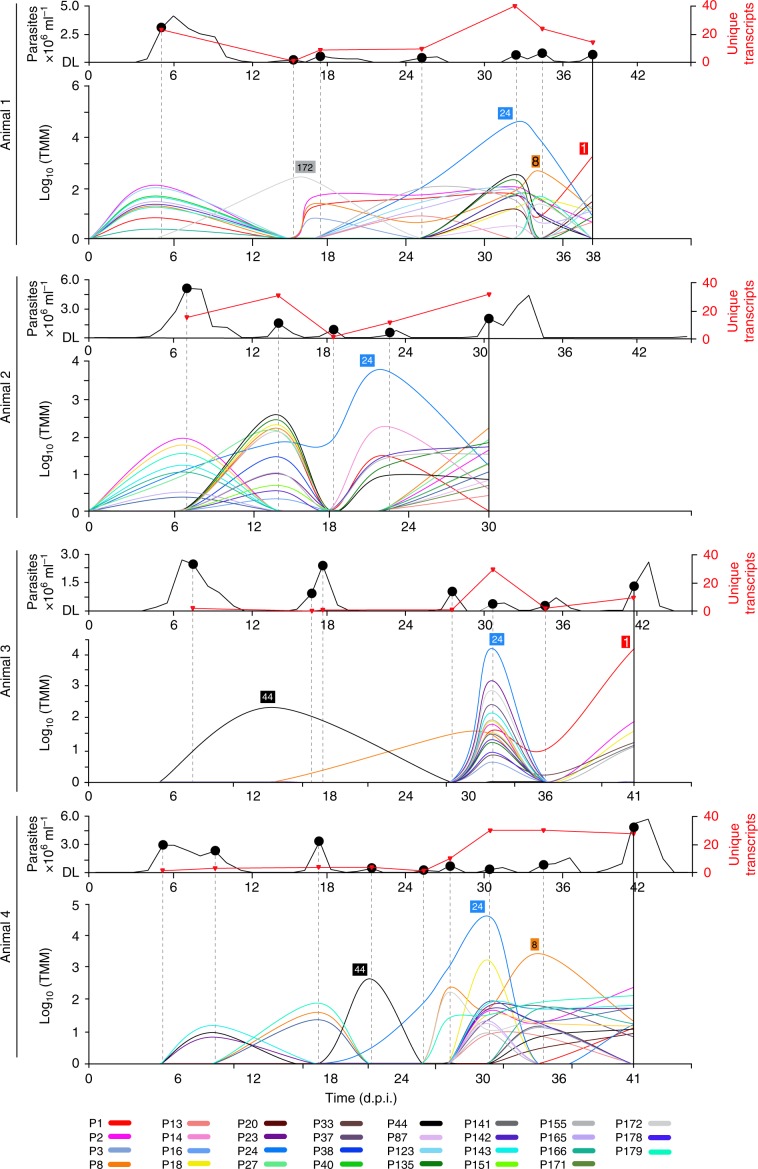
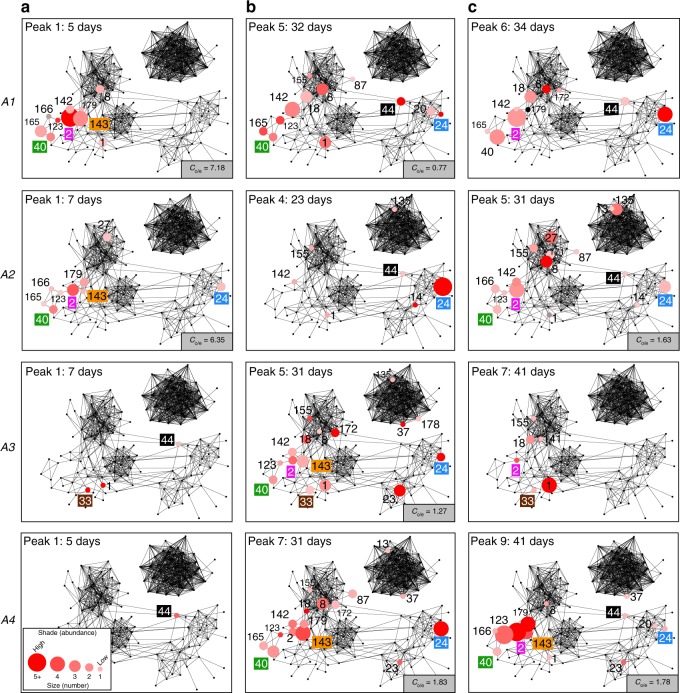
African trypanosomes (Trypanosoma) are vector-borne haemoparasites that survive in the vertebrate bloodstream through antigenic variation of their Variant Surface Glycoprotein (VSG). Recombination, or rather segmented gene conversion, is fundamental in Trypanosoma brucei for both VSG gene switching and for generating antigenic diversity during infections. Trypanosoma vivax is a related, livestock pathogen whose VSG lack structures that facilitate gene conversion in T. brucei and mechanisms underlying its antigenic diversity are poorly understood. Here we show that species-wide VSG repertoire is broadly conserved across diverse T. vivax clinical strains and has limited antigenic repertoire. We use variant antigen profiling, coalescent approaches and experimental infections to show that recombination plays little role in diversifying T. vivax VSG sequences. These results have immediate consequences for both the current mechanistic model of antigenic variation in African trypanosomes and species differences in virulence and transmission, requiring reconsideration of the wider epidemiology of animal African trypanosomiasis.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Antigenic diversity is generated by distinct evolutionary mechanisms in African trypanosome species.Proc Natl Acad Sci U S A. 2012 Feb 28;109(9):3416-21. doi: 10.1073/pnas.1117313109. Epub 2012 Feb 13. Proc Natl Acad Sci U S A. 2012. PMID: 22331916 Free PMC article.
-
Monoallelic expression and epigenetic inheritance sustained by a Trypanosoma brucei variant surface glycoprotein exclusion complex.Nat Commun. 2019 Jul 9;10(1):3023. doi: 10.1038/s41467-019-10823-8. Nat Commun. 2019. PMID: 31289266 Free PMC article.
-
Quantitative sequencing confirms VSG diversity as central to immune evasion by Trypanosoma brucei.Trends Parasitol. 2015 Aug;31(8):346-9. doi: 10.1016/j.pt.2015.05.001. Epub 2015 May 18. Trends Parasitol. 2015. PMID: 25999027 Free PMC article.
-
Escaping the immune system by DNA repair and recombination in African trypanosomes.Open Biol. 2019 Nov 29;9(11):190182. doi: 10.1098/rsob.190182. Epub 2019 Nov 13. Open Biol. 2019. PMID: 31718509 Free PMC article. Review.
-
A Host-Pathogen Interaction Reduced to First Principles: Antigenic Variation in T. brucei.Results Probl Cell Differ. 2015;57:23-46. doi: 10.1007/978-3-319-20819-0_2. Results Probl Cell Differ. 2015. PMID: 26537376 Review.
Cited by
-
Transcriptomic profiling of Trypanosoma congolense mouthpart parasites from naturally infected flies.Parasit Vectors. 2022 May 2;15(1):152. doi: 10.1186/s13071-022-05258-y. Parasit Vectors. 2022. PMID: 35501882 Free PMC article.
-
The Glycosylphosphatidylinositol Anchor: A Linchpin for Cell Surface Versatility of Trypanosomatids.Front Cell Dev Biol. 2021 Nov 1;9:720536. doi: 10.3389/fcell.2021.720536. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34790656 Free PMC article. Review.
-
Vivaxin genes encode highly immunogenic, non-variant antigens on the Trypanosoma vivax cell-surface.PLoS Negl Trop Dis. 2022 Sep 21;16(9):e0010791. doi: 10.1371/journal.pntd.0010791. eCollection 2022 Sep. PLoS Negl Trop Dis. 2022. PMID: 36129968 Free PMC article.
-
Investigation of Trypanosoma-induced vascular damage sheds insights into Trypanosoma vivax sequestration.Cell Surf. 2023 Oct 27;10:100113. doi: 10.1016/j.tcsw.2023.100113. eCollection 2023 Dec 15. Cell Surf. 2023. PMID: 37954640 Free PMC article.
-
Evasive mechanisms of human VSG and PfEMP1 antigens with link to Vaccine scenario: a review.J Parasit Dis. 2025 Mar;49(1):13-28. doi: 10.1007/s12639-024-01740-9. Epub 2024 Sep 24. J Parasit Dis. 2025. PMID: 39975623 Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
