

ATM and PRDM9 regulate SPO11-bound recombination intermediates during meiosis
- PMID: 32051414
- PMCID: PMC7016097
- DOI: 10.1038/s41467-020-14654-w
ATM and PRDM9 regulate SPO11-bound recombination intermediates during meiosis
Abstract
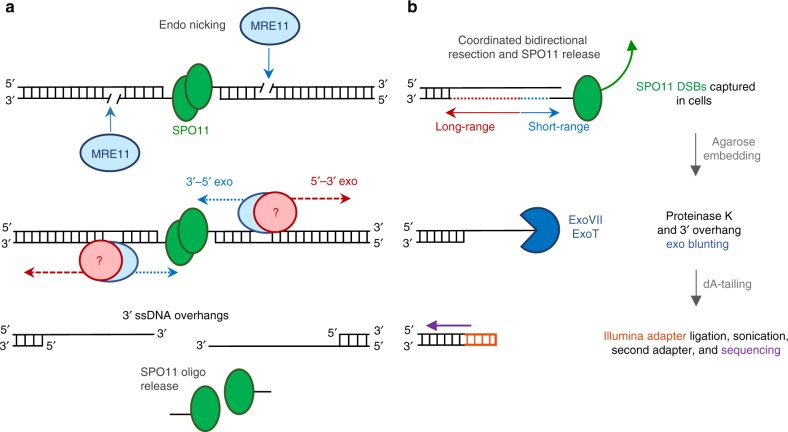
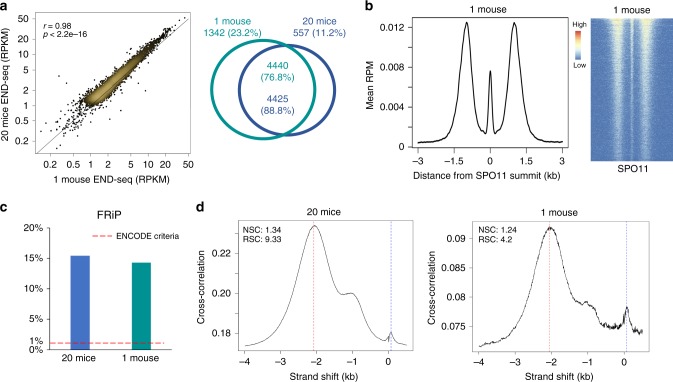
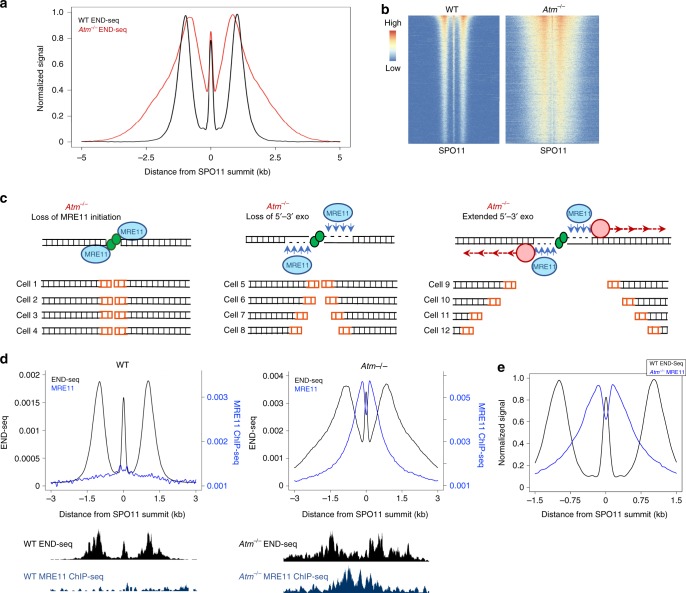
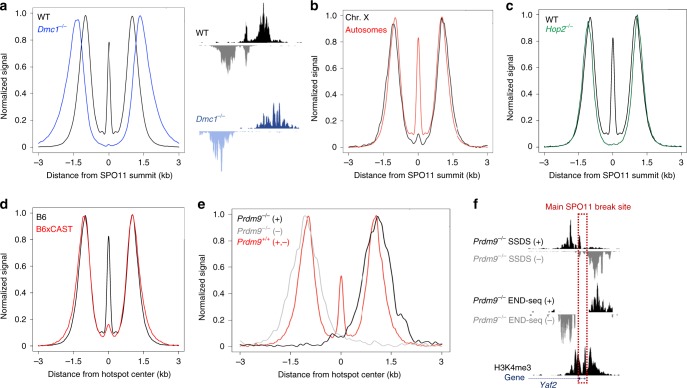
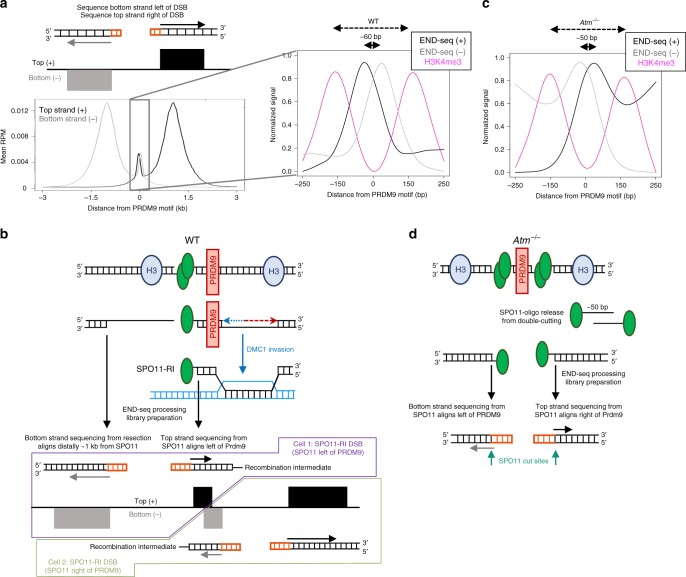
Meiotic recombination is initiated by SPO11-induced double-strand breaks (DSBs). In most mammals, the methyltransferase PRDM9 guides SPO11 targeting, and the ATM kinase controls meiotic DSB numbers. Following MRE11 nuclease removal of SPO11, the DSB is resected and loaded with DMC1 filaments for homolog invasion. Here, we demonstrate the direct detection of meiotic DSBs and resection using END-seq on mouse spermatocytes with low sample input. We find that DMC1 limits both minimum and maximum resection lengths, whereas 53BP1, BRCA1 and EXO1 play surprisingly minimal roles. Through enzymatic modifications to END-seq, we identify a SPO11-bound meiotic recombination intermediate (SPO11-RI) present at all hotspots. We propose that SPO11-RI forms because chromatin-bound PRDM9 asymmetrically blocks MRE11 from releasing SPO11. In Atm-/- spermatocytes, trapped SPO11 cleavage complexes accumulate due to defective MRE11 initiation of resection. Thus, in addition to governing SPO11 breakage, ATM and PRDM9 are critical local regulators of mammalian SPO11 processing.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
