

Genetic basis for iMCD-TAFRO
- PMID: 32051554
- PMCID: PMC7148173
- DOI: 10.1038/s41388-020-1204-9
Genetic basis for iMCD-TAFRO
Abstract
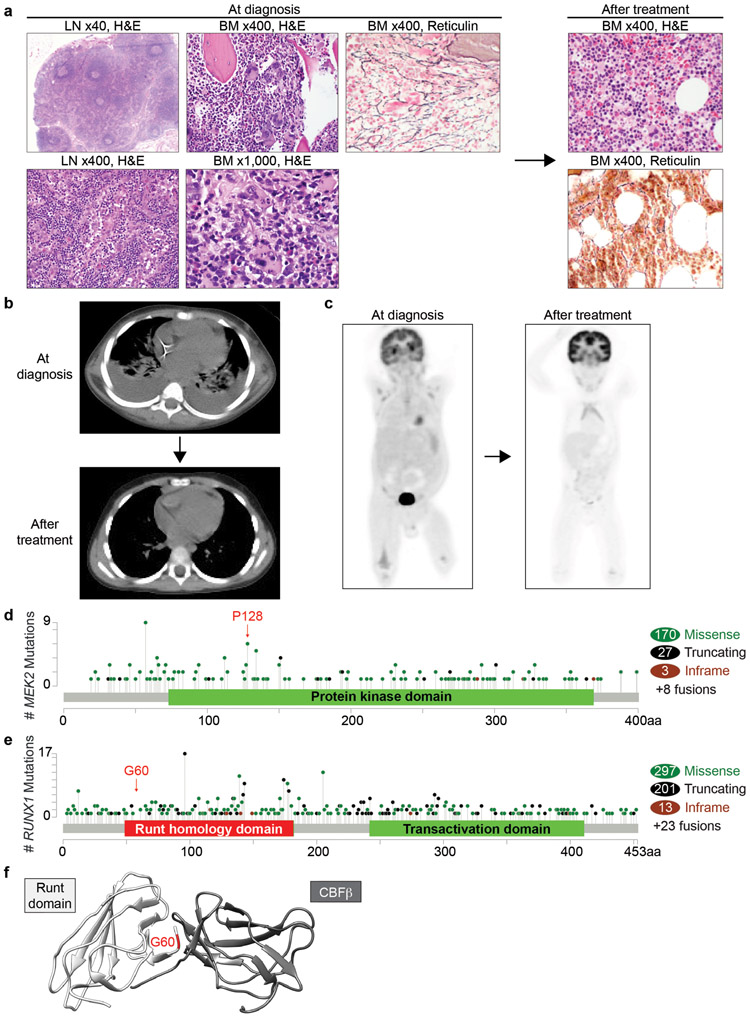
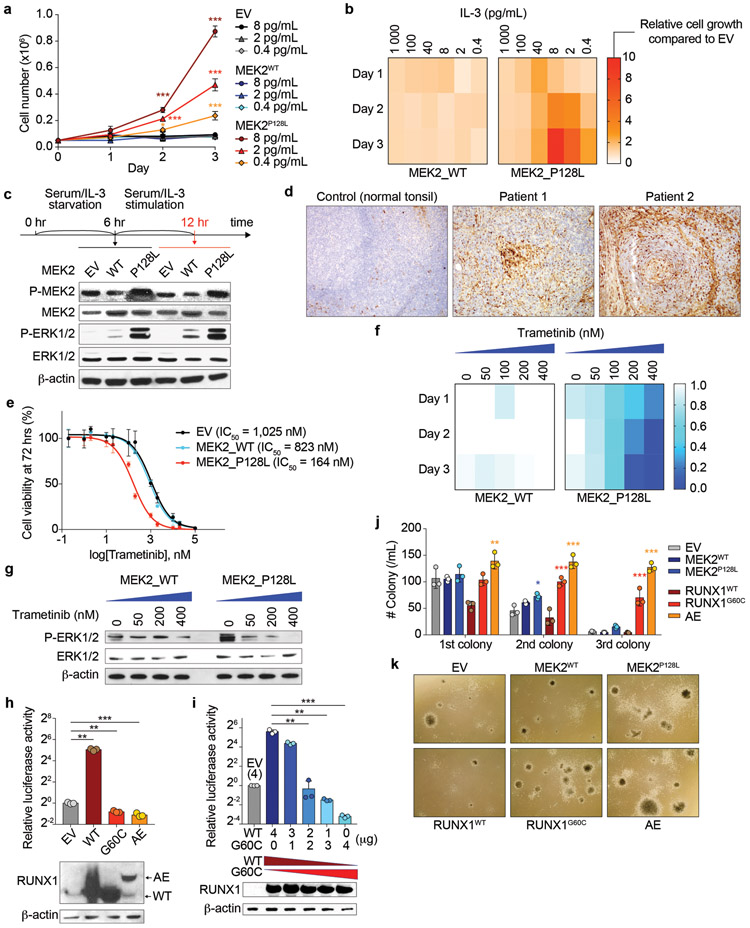
TAFRO syndrome, a clinical subtype of idiopathic multicentric Castleman disease (iMCD), consists of a constellation of symptoms/signs including thrombocytopenia, anasarca, fever, reticulin fibrosis/renal dysfunction, and organomegaly. The etiology of iMCD-TAFRO and the basis for cytokine hypersecretion commonly seen in iMCD-TAFRO patients has not been elucidated. Here, we identified a somatic MEK2P128L mutation and a germline RUNX1G60C mutation in two patients with iMCD-TAFRO, respectively. The MEK2P128L mutation, which has been identified previously in solid tumor and histiocytosis patients, caused hyperactivated MAP kinase signaling, conferred IL-3 hypersensitivity and sensitized the cells to various MEK inhibitors. The RUNX1G60C mutation abolished the transcriptional activity of wild-type RUNX1 and functioned as a dominant negative form of RUNX1, resulting in enhanced self-renewal activity in hematopoietic stem/progenitor cells. Interestingly, ERK was heavily activated in both patients, highlighting a potential role for activation of MAPK signaling in iMCD-TAFRO pathogenesis and a rationale for exploring inhibition of the MAPK pathway as a therapy for iMCD-TAFRO. Moreover, these data suggest that iMCD-TAFRO might share pathogenetic features with clonal inflammatory disorders bearing MEK and RUNX1 mutations such as histiocytoses and myeloid neoplasms.
Conflict of interest statement
Conflict of interest
DCF receives research funding from EUSA Pharma for the ACCELERATE Registry (formerly sponsored by Janssen Pharmaceuticals). A.D. has received personal fees from Roche, Corvus Pharmaceuticals, Physicians’ Education Resource, Seattle Genetics, Peerview Institute, Oncology Specialty Group, Pharmacyclics, Celgene, and Novartis and research grants from National Cancer Institute, Roche. O.A.-W. has served as a consultant for H3 Biomedicine, Foundation Medicine Inc., Merck, and Janssen and serves on the scientific advisory board of Envisagenics Inc.; O.A.-W. has received personal speaking fees from Daiichi Sankyo. O.A.-W. has received prior research funding from H3 Biomedicine unrelated to the current manuscript. O.A.-W. is an inventor on a provisional patent application submitted by Fred Hutchinson Cancer Research Center that covers BRD9 activation in cancer. W.X has received research support from Stemline therapeutics. Other authors have nothing to disclose.
Figures
References
-
- Kawabata H, Takai K, Kojima M, Nakamura N, Aoki S, Nakamura S, et al. Castleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly: a status report and summary of Fukushima (6 June, 2012) and Nagoya meetings (22 September, 2012). J Clin Exp Hematop. 2013;53. - PubMed
-
- Inoue M, Ankou M, Hua J, Iwaki Y, Hagihara M, Ota Y. Complete resolution of TAFRO syndrome (thrombocytopenia, anasarca, fever, reticulin fibrosis and organomegaly) after immunosuppressive therapies using corticosteroids and cyclosporin A : a case report. J Clin Exp Hematop. 2013;53:95–9. - PubMed
-
- Iwaki N, Fajgenbaum DC, Nabel CS, Gion Y, Kondo E, Kawano M, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol. 2016;91:220–6. - PubMed
-
- Pierson SK, Stonestrom AJ, Shilling D, Ruth J, Nabel CS, Singh A, et al. Plasma proteomics identifies a ‘chemokine storm’ in idiopathic multicentric Castleman disease. Am J Hematol. 2018;93:902–12. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
