

doi: 10.1093/eurheartj/ehz962.
Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel
Affiliations
- PMID: 32052833
- PMCID: PMC7308544
- DOI: 10.1093/eurheartj/ehz962
Item in Clipboard
Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel
Eur Heart J.
.
No abstract available
Figures
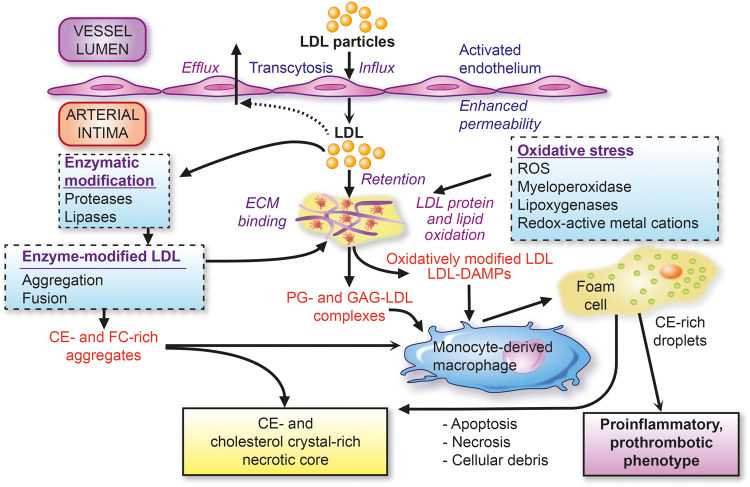
Low-density lipoprotein (LDL) as the primary driver of atherogenesis. Key features of the influx and retention of LDL in the arterial intima, with ensuing pathways of modification leading to (i) extracellular cholesterol accumulation and (ii) formation of cholesteryl ester droplet-engorged macrophage foam cells with transformation to an inflammatory and prothrombotic phenotype. Both of these major pathways favour formation of the plaque necrotic core containing cellular and extracellular debris and LDL-cholesterol-derived cholesterol crystals. CE, cholesteryl ester; DAMPs, damage-associated molecular patterns; ECM, extracellular matrix; FC, free cholesterol; GAG, glycosaminoglycans; PG, proteoglycans; ROS, reactive oxygen species.
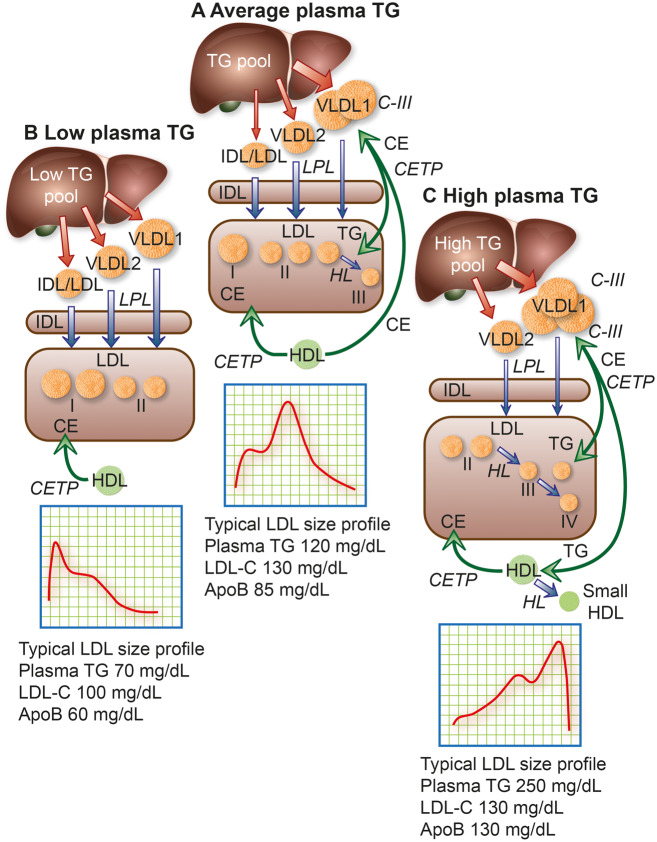
Model of the metabolic interrelationships between low-density lipoprotein (LDL) subfractions and their hepatic precursors. The liver produces apolipoprotein (apo)B100-containing particles ranging in size from large triglyceride (TG)-rich very low-density lipoprotein (VLDL) 1, through small VLDL2 and intermediate-density lipoprotein (IDL) to LDL. The hepatic TG content (TG pool) affects the profile of the secreted particles. Secreted VLDL undergoes lipolysis and remodelling to form remnants/IDL; LDL is then formed via the actions of lipoprotein lipase (LPL), hepatic lipase (HL), and cholesteryl ester transfer protein (CETP). (A) In people with population average TG levels, about half the lipolytic remnants (which correspond to IDL based on density and size) in this pathway are cleared relatively efficiently and the remainder are converted mainly to LDL-II, which has higher LDL receptor affinity and shorter residence time than the LDL arising from VLDL1.,,, The composition of IDL-derived LDL is modulated both by CETP-mediated transfer of cholesteryl esters (CE) from high-density lipoprotein (HDL) and by CETP-mediated transfer of TG from VLDL and their remnants., (B) In individuals with low plasma TG, LDL-I and -II predominate. Clearance of these lipoproteins is rapid and LDL-cholesterol (LDL-C) and apoB concentrations are low. (C) Individuals with elevated plasma TG levels overproduce VLDL1 and have reduced lipolysis rates due in part to inhibition of LPL activity by their abundant content of apoC-III, an LPL inhibitor. Very low-density lipoprotein 1 remodelling gives rise to remnants within the VLDL size range that are enriched in apoE; such circulating remnants can be removed by several mechanisms, primarily in the liver, including the LDL receptor-related protein, heparan sulfate proteoglycans, and LDL receptor. Hepatic clearance of VLDL1-derived remnant particles may, however, be slowed by enrichment with apoC-III. Very low-density lipoprotein 1 and VLDL2 are targeted by CETP, which exchanges core CE in LDL for TG in both VLDL1 and VLDL2. Hydrolysis of TG by HL action then shrinks LDL particles to preferentially form small, dense LDL-III in moderate hypertriglyceridaemia, or even smaller LDL-IV in severe hypertriglyceridaemia; such small dense LDL exhibit attenuated binding affinity for the LDL receptor, resulting in prolonged plasma residence (Box ). Together, this constellation of lipoprotein changes, originating in increased levels of large VLDL1 and small dense LDL, represents a lipid phenotype designated atherogenic dyslipidaemia,,,,, a key feature of metabolic syndrome and Type 2 diabetes., Typical LDL subfraction patterns are indicated together with relevant plasma lipid and apoB levels. Note that when small dense LDL is abundant, apoB is elevated more than LDL-C. The width of the red arrows reflects the quantity of apoB/particle production and release from the liver, while the width of the blue arrows depicts relative lipolytic efficiency.
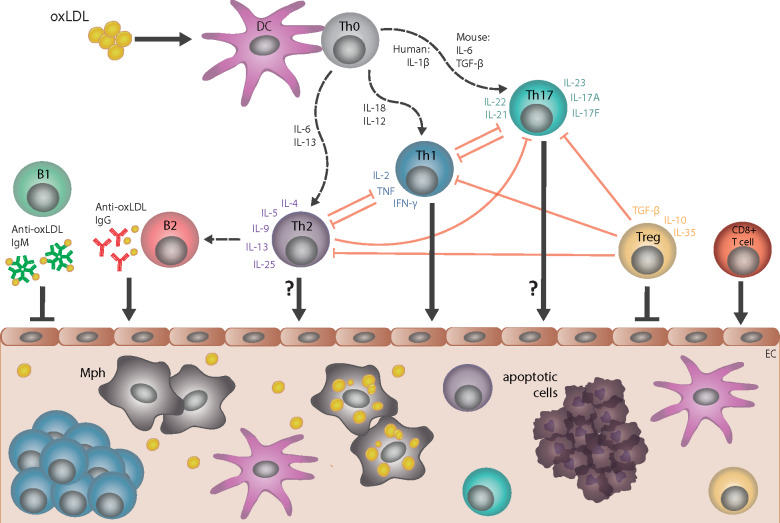
Cellular and humoral immune responses in atherosclerosis. Dendritic cells (DC) take up several forms of modified low-density lipoprotein (LDL), including oxidized LDL (oxLDL), and present specific epitopes (e.g. apolipoprotein B peptides) to naive T cells (Th0), which induces differentiation into CD4+ T helper 1 (Th1), T helper 2 (Th2), T helper 17 (Th17), or T regulatory (T reg) cell subtypes; multiple cytokines control such differentiation. CD4+ T-cell subtypes, together with specific cytokines that they secrete, provide help to B cells and regulate the activity of other T-cell subtypes. The pro-atherogenic role of interferon gamma (IFN-γ)-secreting Th1 cells and the anti-atherogenic effect of interleukin-10/transforming growth factor beta (IL-10/TGF-β)-secreting T regulatory cells are well established. However, the role of Th2 and Th17 in atherogenesis is less clear, as opposing effects of cytokines associated with these respective subtypes have been described. Cytotoxic CD8+ T cells can promote atherogenesis. Anti-oxLDL immunoglobulin (Ig)M antibodies produced by B1 cells are atheroprotective, whereas anti-oxLDL IgG antibodies produced by B2-cell subsets are likely pro-atherogenic. All of these cell types may infiltrate the arterial wall at sites of ongoing plaque development, with the possible exception of Th2 and Th17 cell types. EC, endothelial cell; Mph, monocyte-derived macrophage.
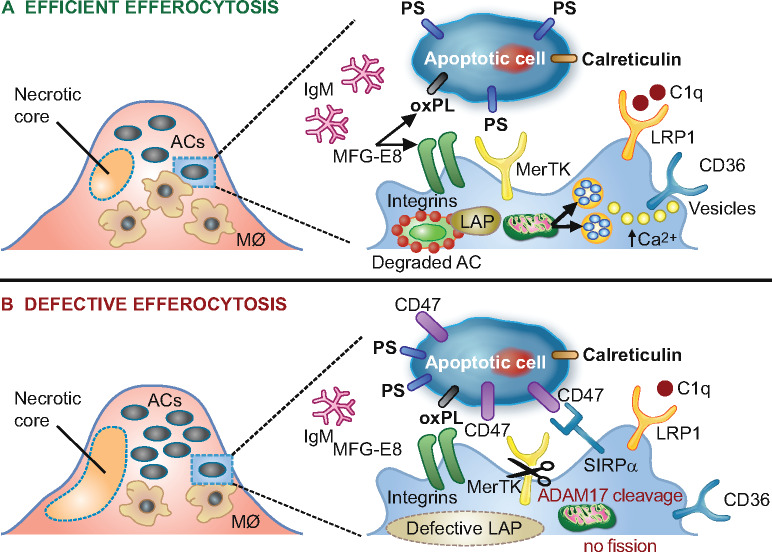
Schematic representation of processes involved in lesional efferocytosis. (A) Externalized ‘eat me’ signals including phosphatidylserine (PS), calreticulin, and oxidized phospholipids (oxPL) are recognized by their respective receptors, Mer tyrosine kinase (MerTK), low-density lipoprotein-receptor-related protein 1 (LRP1), as well as integrin αvβ3 and CD36 on macrophages; such recognition is facilitated either directly or mediated by bridging molecules such as growth arrest-specific 6 for PS, complement protein C1q for calreticulin and milk fat globule-epidermal growth factor 8 (MFG-E8) for oxPL. Calcium-dependent vesicular trafficking events driven by mitochondrial fission and LC3-associated phagocytosis (LAP) promote phagolysosomal fusion and the hydrolytic degradation of apoptotic cells. Simultaneously, natural immunoglobulin (Ig)M antibodies with reactivity towards oxidation-specific epitopes further enhance the efficient clearance of dying cells via complement receptors. (B) In advanced atherosclerosis, one or more of these mechanisms are dysfunctional and can lead to defective efferocytosis, propagating non-resolving inflammation and plaque necrosis. Additional processes contributing to impaired efferocytosis include ADAM-17-mediated cleavage of MerTK as well as the inappropriate expression of the ‘don’t eat me’ signal CD47 on apoptotic cell surfaces. ACs, apoptotic cells.
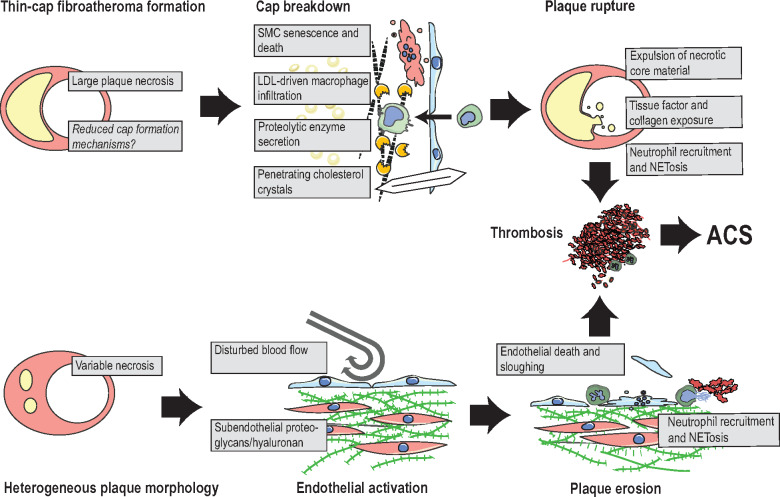
Proposed mechanisms of plaque rupture and plaque erosion. Rupture: lesions that develop extensive necrosis and only sparse fibrous cap tissue are at risk of plaque rupture. Suggested final processes that precipitate rupture include senescence and death of residual cap smooth muscle cells (SMC), degradation of the fibrous matrix by macrophage-secreted proteolytic enzymes, and cholesterol crystals, which may penetrate cap tissue. These processes expose the prothrombotic plaque interior and result in neutrophil-accelerated thrombosis. Erosion: lesions that are complicated by erosion typically display variable amounts of plaque necrosis, but are frequently characterized by subendothelial accumulation of proteoglycans and hyaluronan. Current hypotheses suggest that the combination of disturbed blood flow and endothelial activation by immune activators, e.g. hyaluronan fragments, leads to neutrophil recruitment with neutrophil extracellular trapsosis, endothelial cell apoptosis/sloughing, and thrombus formation. ACS, acute coronary syndrome; NETosis, cell death by neutrophil extracellular traps.
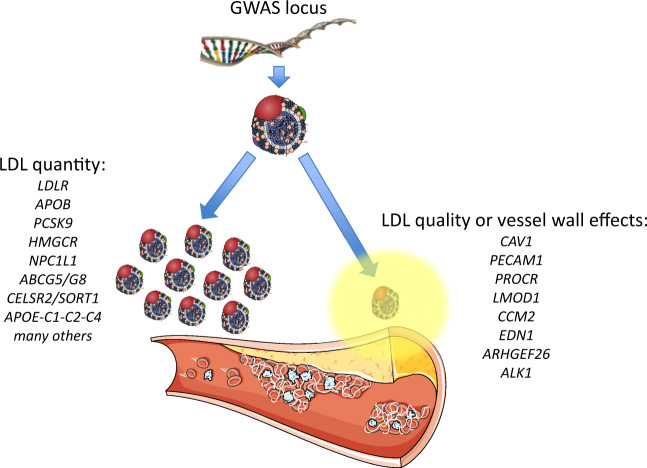
Genomic loci associated with atherosclerosis. Loci identified by genome-wide association studies (GWAS) can have different effects on low-density lipoprotein (LDL). On the left are shown selected GWAS loci associated with LDL-cholesterol (LDL-C) levels, several of which are associated with atherosclerosis events and are incorporated in predictive risk scores. Many have also been independently validated in Mendelian randomization studies and in studies of rare families. Some are proven drug targets to reduce clinical events. On the right are shown loci that do not primarily affect LDL-C levels, but may instead underlie qualitative changes in either the particle itself or in the vessel wall to locally promote atherogenesis.
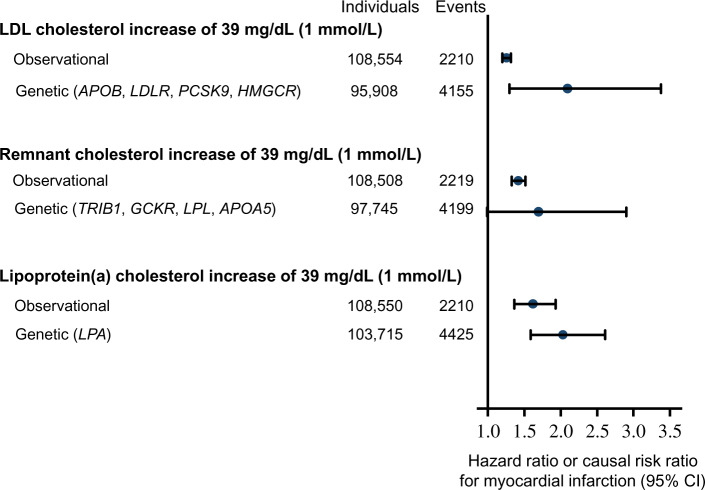
Comparison of risk of myocardial infarction by 1 mmol/L (39 mg/dL) higher levels of low-density lipoprotein (LDL) cholesterol, remnant-cholesterol, and lipoprotein(a)-cholesterol from observational and genetic studies. Data from individuals in the Copenhagen General Population Study adapted with permission from Nordestgaard et al.
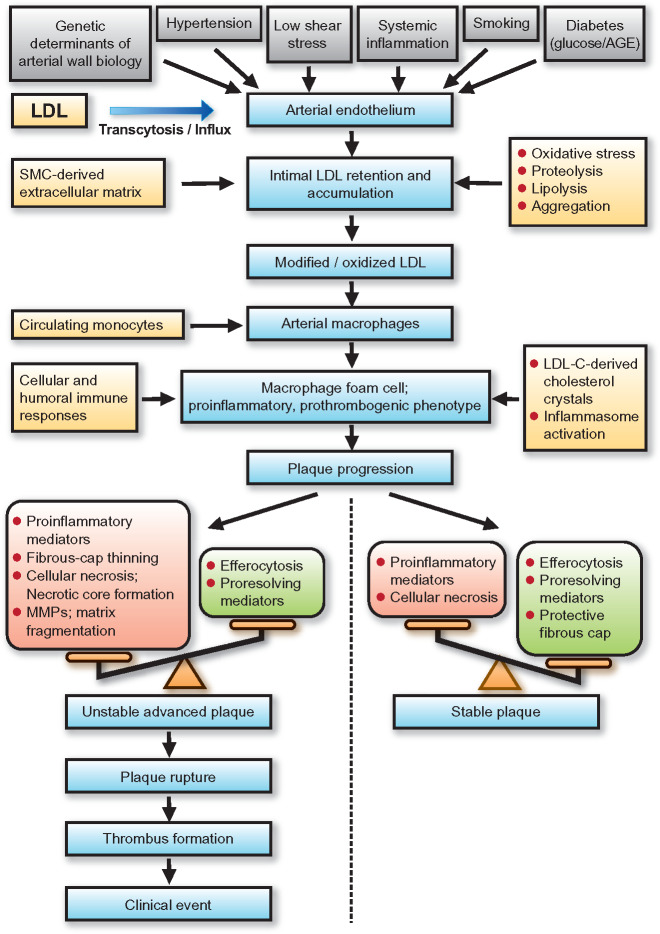
Low-density lipoprotein (LDL) and atherobiology. Summary of the principal mechanisms underlying the entry, retention, and accumulation of LDL particles in the artery wall, and subsequent LDL-driven downstream events that are central to the complex pathogenesis of atherothrombosis. Intermediate fatty streak lesions are characterized by subintimal accumulation of macrophage foam cells. AGE, advanced glcation end-products; LDL-C, LDL-cholesterol; MMPs, matrix metallopeptidases
References
-
- Berenson GS, Srinivasan SR, Bao W, Newman WP 3rd, Tracy RE, Wattigney WA.. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med 1998;338:1650–1656. - PubMed
-
- Newman WP 3rd, Freedman DS, Voors AW, Gard PD, Srinivasan SR, Cresanta JL, Williamson GD, Webber LS, Berenson GS.. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa Heart Study. N Engl J Med 1986;314:138–144. - PubMed
-
- Fernandez-Friera L, Penalvo JL, Fernandez-Ortiz A, Ibanez B, Lopez-Melgar B, Laclaustra M, Oliva B, Mocoroa A, Mendiguren J, Martinez de Vega V, Garcia L, Molina J, Sanchez-Gonzalez J, Guzman G, Alonso-Farto JC, Guallar E, Civeira F, Sillesen H, Pocock S, Ordovas JM, Sanz G, Jimenez-Borreguero LJ, Fuster V.. Prevalence, vascular distribution, and multiterritorial extent of subclinical atherosclerosis in a middle-aged cohort: the PESA (Progression of Early Subclinical Atherosclerosis) study. Circulation 2015;131:2104–2113. - PubMed
-
- Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, Watts GF, Borén J, Fazio S, Horton JD, Masana L, Nicholls SJ, Nordestgaard BG, van de Sluis B, Taskinen M-R, Tokgözoğlu L, Landmesser U, Laufs U, Wiklund O, Stock JK, Chapman MJ, Catapano AL.. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J 2017;38:2459–2472. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
