

Antibiotic resistance prediction for Mycobacterium tuberculosis from genome sequence data with Mykrobe
- PMID: 32055708
- PMCID: PMC7004237
- DOI: 10.12688/wellcomeopenres.15603.1
Antibiotic resistance prediction for Mycobacterium tuberculosis from genome sequence data with Mykrobe
Abstract
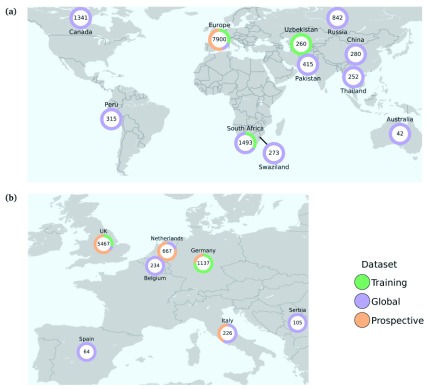
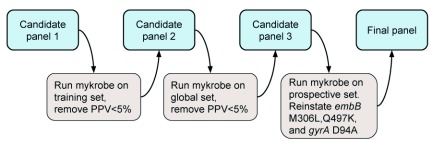
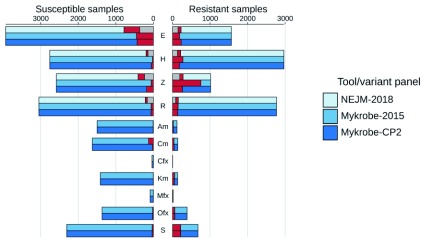
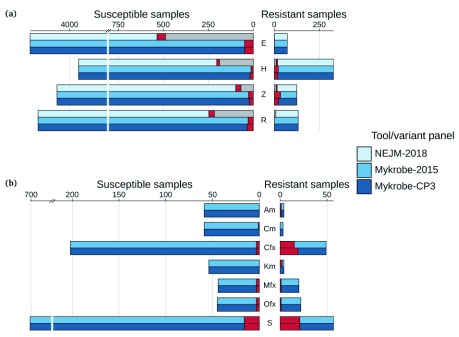
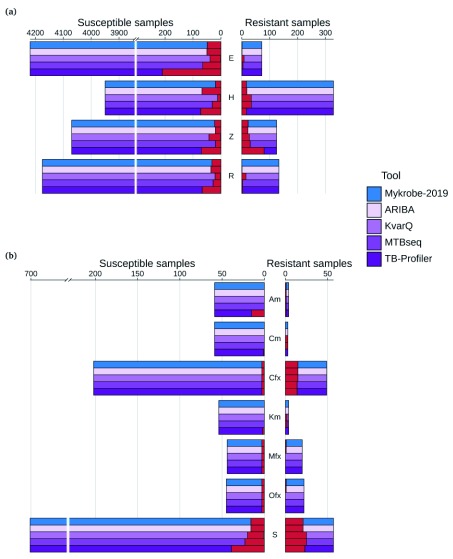
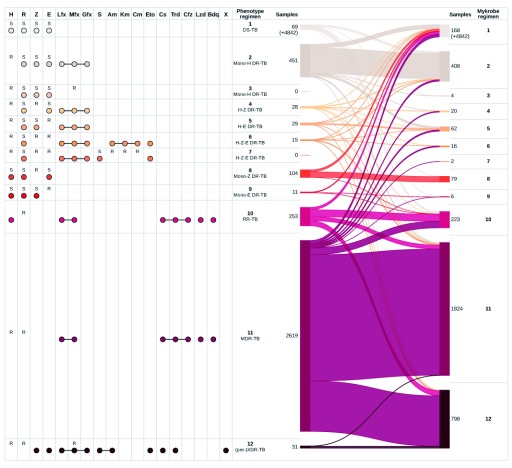
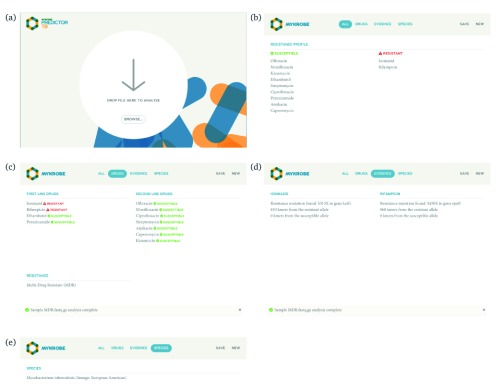
Two billion people are infected with Mycobacterium tuberculosis, leading to 10 million new cases of active tuberculosis and 1.5 million deaths annually. Universal access to drug susceptibility testing (DST) has become a World Health Organization priority. We previously developed a software tool, Mykrobe predictor, which provided offline species identification and drug resistance predictions for M. tuberculosis from whole genome sequencing (WGS) data. Performance was insufficient to support the use of WGS as an alternative to conventional phenotype-based DST, due to mutation catalogue limitations. Here we present a new tool, Mykrobe, which provides the same functionality based on a new software implementation. Improvements include i) an updated mutation catalogue giving greater sensitivity to detect pyrazinamide resistance, ii) support for user-defined resistance catalogues, iii) improved identification of non-tuberculous mycobacterial species, and iv) an updated statistical model for Oxford Nanopore Technologies sequencing data. Mykrobe is released under MIT license at https://github.com/mykrobe-tools/mykrobe. We incorporate mutation catalogues from the CRyPTIC consortium et al. (2018) and from Walker et al. (2015), and make improvements based on performance on an initial set of 3206 and an independent set of 5845 M. tuberculosis Illumina sequences. To give estimates of error rates, we use a prospectively collected dataset of 4362 M. tuberculosis isolates. Using culture based DST as the reference, we estimate Mykrobe to be 100%, 95%, 82%, 99% sensitive and 99%, 100%, 99%, 99% specific for rifampicin, isoniazid, pyrazinamide and ethambutol resistance prediction respectively. We benchmark against four other tools on 10207 (=5845+4362) samples, and also show that Mykrobe gives concordant results with nanopore data. We measure the ability of Mykrobe-based DST to guide personalized therapeutic regimen design in the context of complex drug susceptibility profiles, showing 94% concordance of implied regimen with that driven by phenotypic DST, higher than all other benchmarked tools.
Keywords: Antimicrobial resistance; antibiotic treatment; diagnostic; nanopore; tuberculosis; whole genome sequencing.
Copyright: © 2019 Hunt M et al.
Conflict of interest statement
No competing interests were disclosed.
Figures
References
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
