

Evolutionary analysis of the most polymorphic gene family in falciparum malaria
- PMID: 32055709
- PMCID: PMC7001760
- DOI: 10.12688/wellcomeopenres.15590.1
Evolutionary analysis of the most polymorphic gene family in falciparum malaria
Abstract
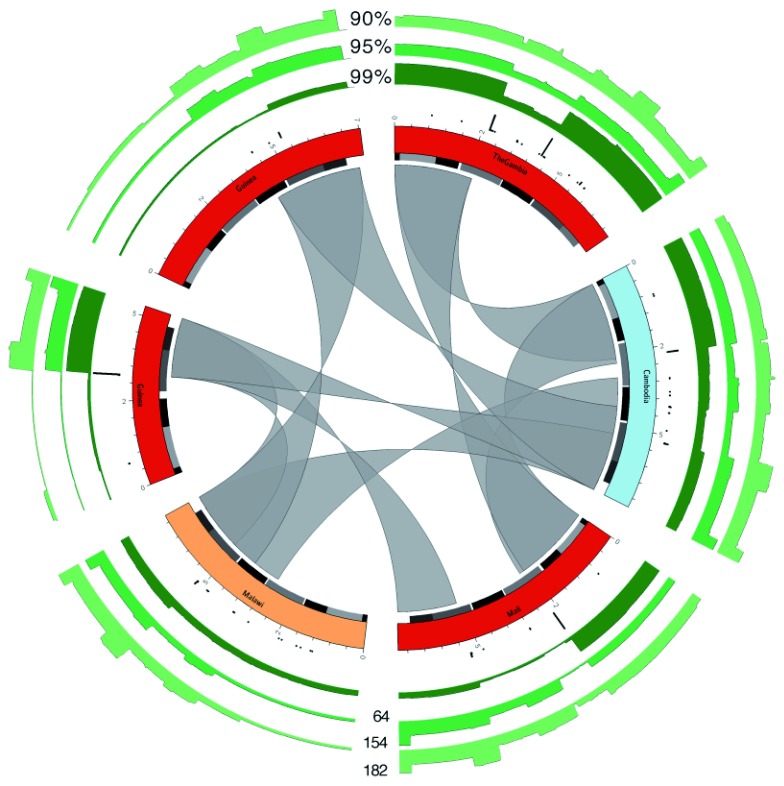
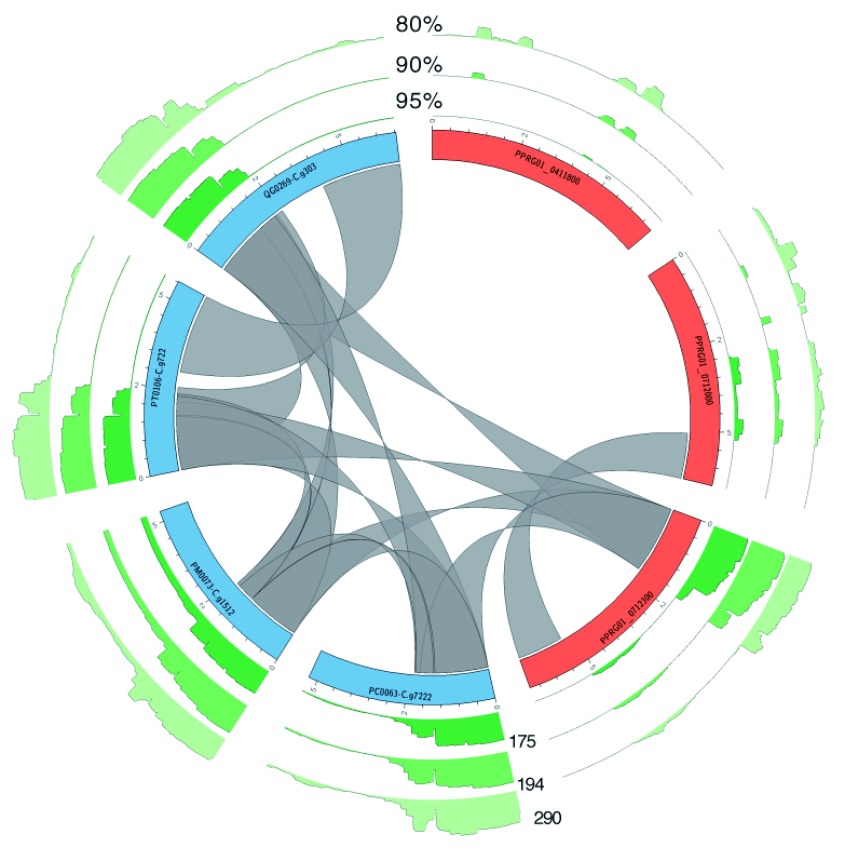
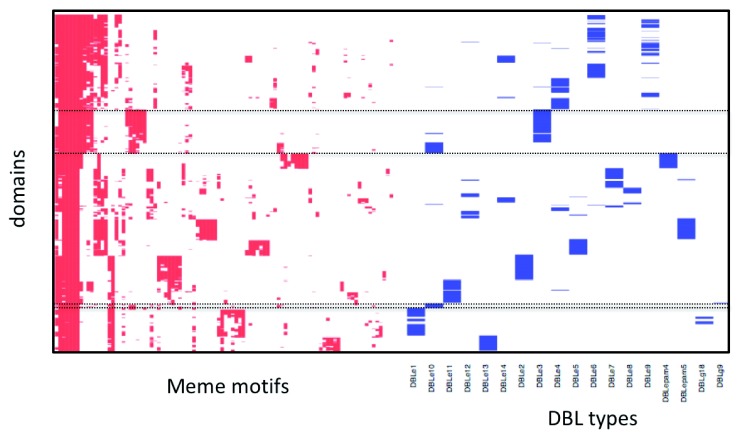
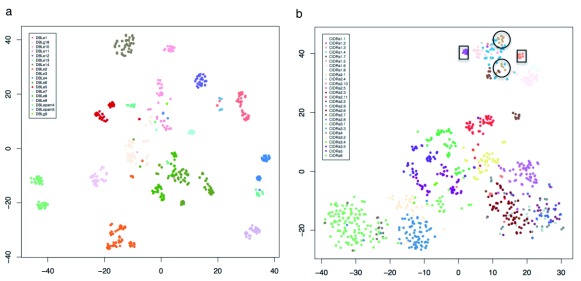
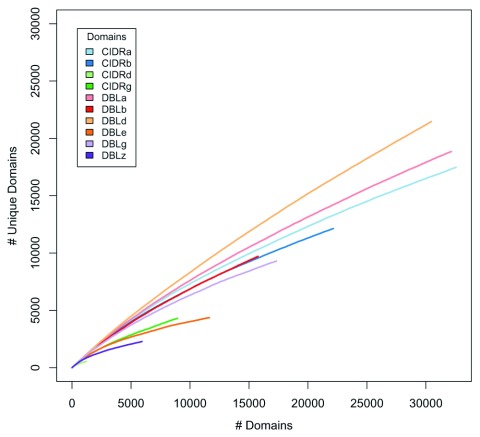
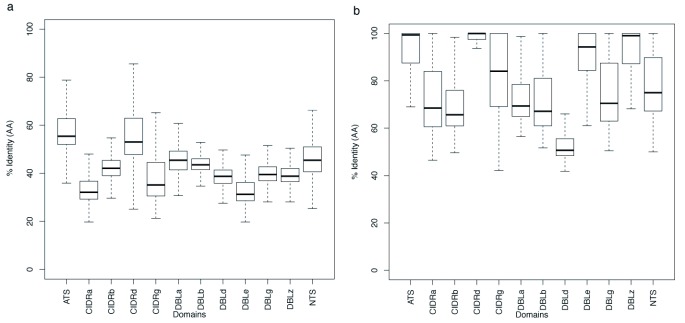
The var gene family of the human malaria parasite Plasmodium falciparum encode proteins that are crucial determinants of both pathogenesis and immune evasion and are highly polymorphic. Here we have assembled nearly complete var gene repertoires from 2398 field isolates and analysed a normalised set of 714 from across 12 countries. This therefore represents the first large scale attempt to catalogue the worldwide distribution of var gene sequences We confirm the extreme polymorphism of this gene family but also demonstrate an unexpected level of sequence sharing both within and between continents. We show that this is likely due to both the remnants of selective sweeps as well as a worrying degree of recent gene flow across continents with implications for the spread of drug resistance. We also address the evolution of the var repertoire with respect to the ancestral genes within the Laverania and show that diversity generated by recombination is concentrated in a number of hotspots. An analysis of the subdomain structure indicates that some existing definitions may need to be revised From the analysis of this data, we can now understand the way in which the family has evolved and how the diversity is continuously being generated. Finally, we demonstrate that because the genes are distributed across the genome, sequence sharing between genotypes acts as a useful population genetic marker.
Keywords: Plasmodium; evolution; var.
Copyright: © 2019 Otto TD et al.
Conflict of interest statement
No competing interests were disclosed.
Figures
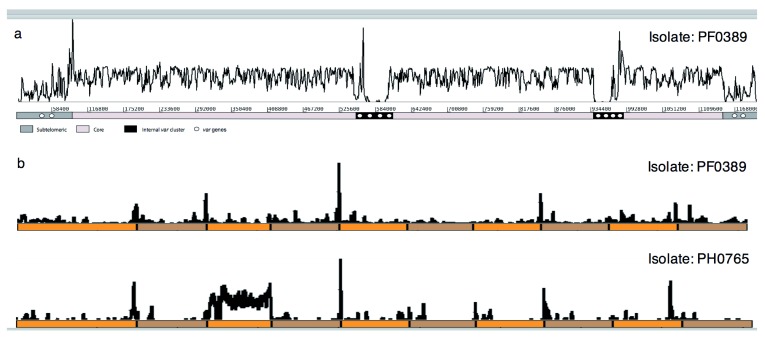
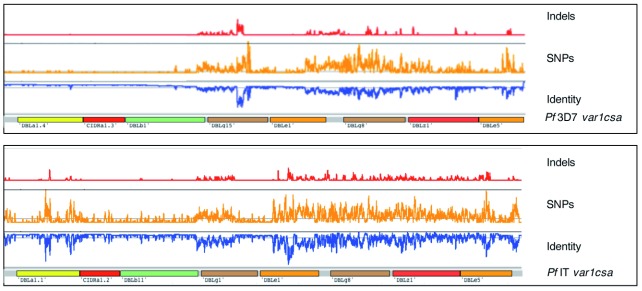
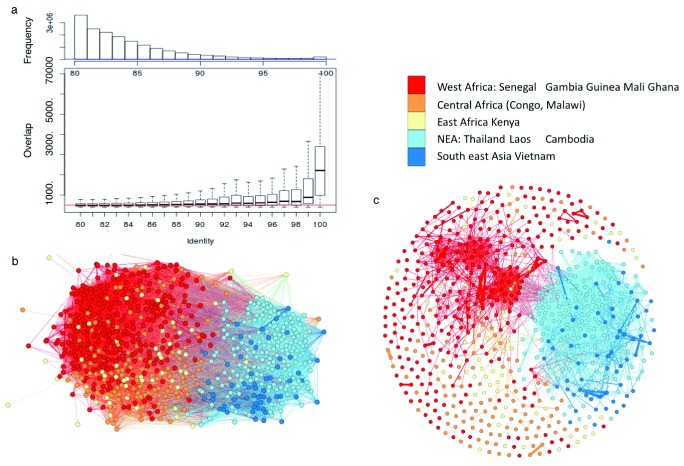
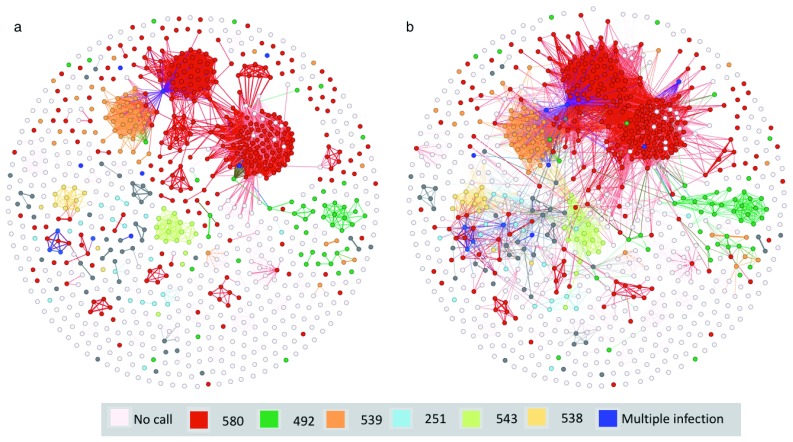
References
Grants and funding
LinkOut - more resources
Full Text Sources
