

Integrative comparison of the genomic and transcriptomic landscape between prostate cancer patients of predominantly African or European genetic ancestry
- PMID: 32059012
- PMCID: PMC7046294
- DOI: 10.1371/journal.pgen.1008641
Integrative comparison of the genomic and transcriptomic landscape between prostate cancer patients of predominantly African or European genetic ancestry
Abstract
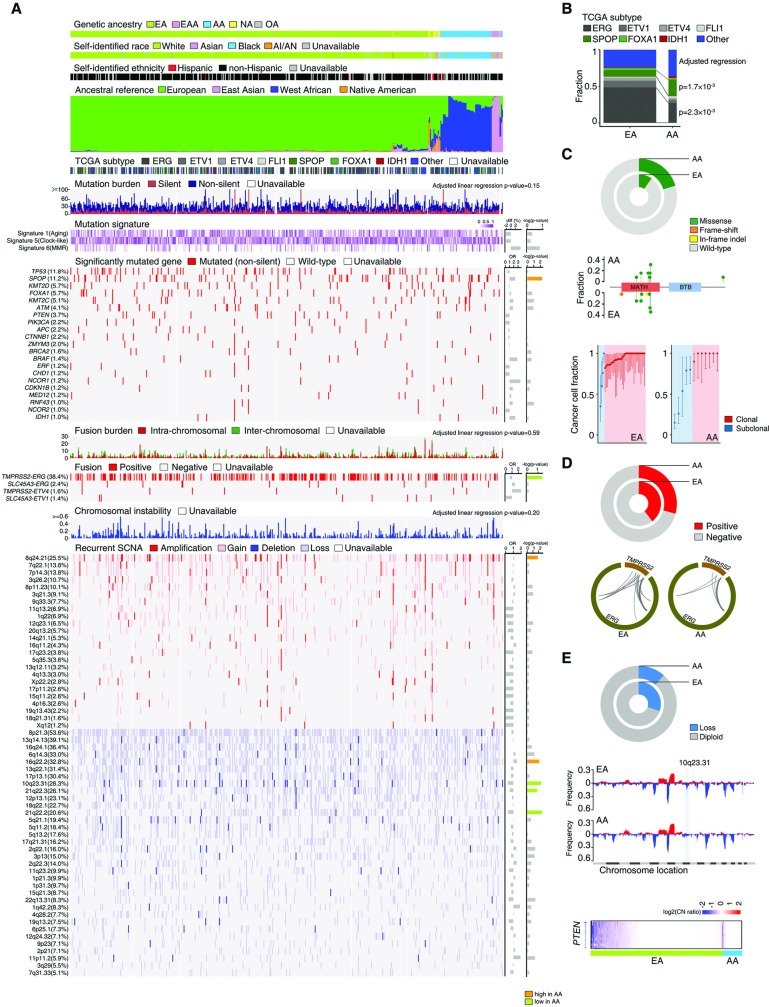
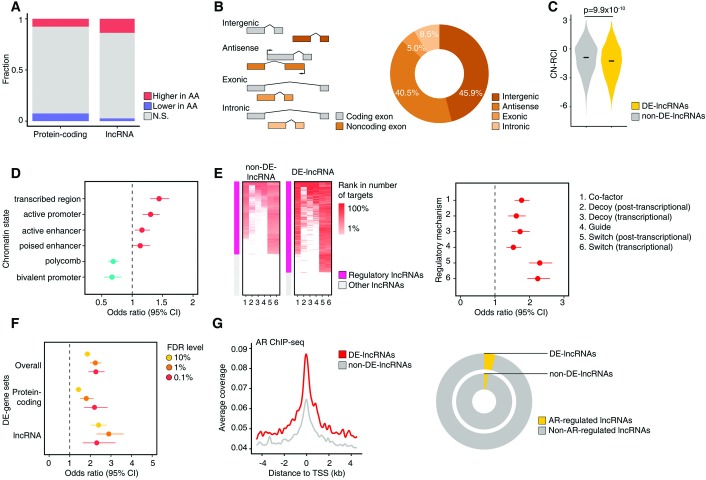
Men of predominantly African Ancestry (AA) have higher prostate cancer (CaP) incidence and worse survival than men of predominantly European Ancestry (EA). While socioeconomic factors drive this disparity, genomic factors may also contribute to differences in the incidence and mortality rates. To compare the prevalence of prostate tumor genomic alterations and transcriptomic profiles by patient genetic ancestry, we evaluated genomic profiles from The Cancer Genome Atlas (TCGA) CaP cohort (n = 498). Patient global and local genetic ancestry were estimated by computational algorithms using genotyping data; 414 (83.1%) were EA, 61 (12.2%) were AA, 11 (2.2%) were East Asian Ancestry (EAA), 10 (2.0%) were Native American (NA), and 2 (0.4%) were other ancestry. Genetic ancestry was highly concordant with self-identified race/ethnicity. Subsequent analyses were limited to 61 AA and 414 EA cases. Significant differences were observed by ancestry in the frequency of SPOP mutations (20.3% AA vs. 10.0% EA; p = 5.6×10-03), TMPRSS2-ERG fusions (29.3% AA vs. 39.6% EA; p = 4.4×10-02), and PTEN deletions/losses (11.5% AA vs. 30.2% EA; p = 3.5×10-03). Differentially expressed genes (DEGs) between AAs and EAs showed significant enrichment for prostate eQTL target genes (p = 8.09×10-48). Enrichment of highly expressed DEGs for immune-related pathways was observed in AAs, and for PTEN/PI3K signaling in EAs. Nearly one-third of DEGs (31.3%) were long non-coding RNAs (DE-lncRNAs). The proportion of DE-lncRNAs with higher expression in AAs greatly exceeded that with lower expression in AAs (p = 1.2×10-125). Both ChIP-seq and RNA-seq data suggested a stronger regulatory role for AR signaling pathways in DE-lncRNAs vs. non-DE-lncRNAs. CaP-related oncogenic lncRNAs, such as PVT1, PCAT1 and PCAT10/CTBP1-AS, were found to be more highly expressed in AAs. We report substantial heterogeneity in the prostate tumor genome and transcriptome between EA and AA. These differences may be biological contributors to racial disparities in CaP incidence and outcomes.
Conflict of interest statement
I have read the journal's policy and the authors of this manuscript have the following competing interests: Drs. Lin Zhang and Xiaowen Hu received research grant from Bristol-Myers Squibb/Celgene. The other authors declare no potential conflicts of interest.
Figures



References
-
- Zeng C, Wen W, Morgans AK, Pao W, Shu XO, Zheng W. Disparities by Race, Age, and Sex in the Improvement of Survival for Major Cancers: Results From the National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program in the United States, 1990 to 2010. JAMA Oncol. 2015;1(1):88–96. 10.1001/jamaoncol.2014.161 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
