

Small airways pathology in idiopathic pulmonary fibrosis: a retrospective cohort study
- PMID: 32061334
- PMCID: PMC7292784
- DOI: 10.1016/S2213-2600(19)30356-X
Small airways pathology in idiopathic pulmonary fibrosis: a retrospective cohort study
Abstract
Background: The observation that patients with idiopathic pulmonary fibrosis (IPF) can have higher than normal expiratory flow rates at low lung volumes led to the conclusion that the airways are spared in IPF. This study aimed to re-examine the hypothesis that airways are spared in IPF using a multiresolution imaging protocol that combines multidetector CT (MDCT), with micro-CT and histology.
Methods: This was a retrospective cohort study comparing explanted lungs from patients with severe IPF treated by lung transplantation with a cohort of unused donor (control) lungs. The donor control lungs had no known lung disease, comorbidities, or structural lung injury, and were deemed appropriate for transplantation on review of the clinical files. The diagnosis of IPF in the lungs from patients was established by a multidisciplinary consensus committee according to existing guidelines, and was confirmed by video-assisted thoracic surgical biopsy or by pathological examination of the contralateral lung. The control and IPF groups were matched for age, sex, height, and bodyweight. Samples of lung tissue were compared using the multiresolution imaging approach: a cascade of clinical MDCT, micro-CT, and histological imaging. We did two experiments: in experiment 1, all the lungs were randomly sampled; in experiment 2, samples were selected from regions of minimal and established fibrosis. The patients and donors were recruited from the Katholieke Universiteit Leuven (Leuven, Belgium) and the University of Pennsylvania Hospital (Philadelphia, PA, USA). The study took place at the Katholieke Universiteit Leuven, and the University of British Columbia (Vancouver, BC, Canada).
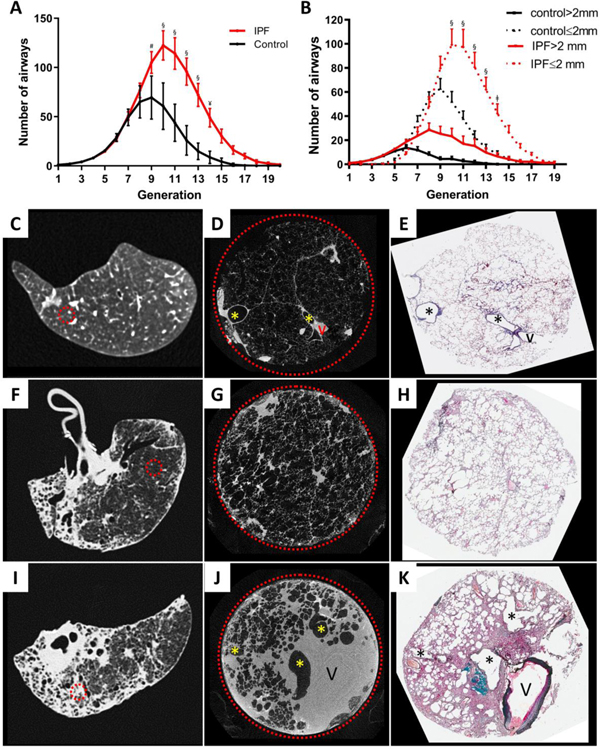
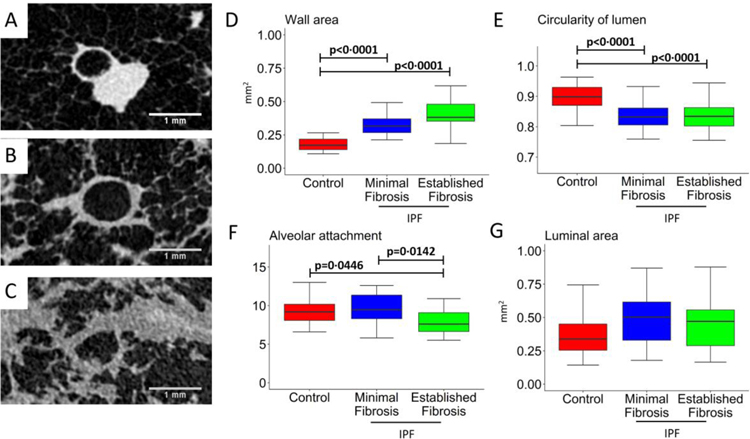
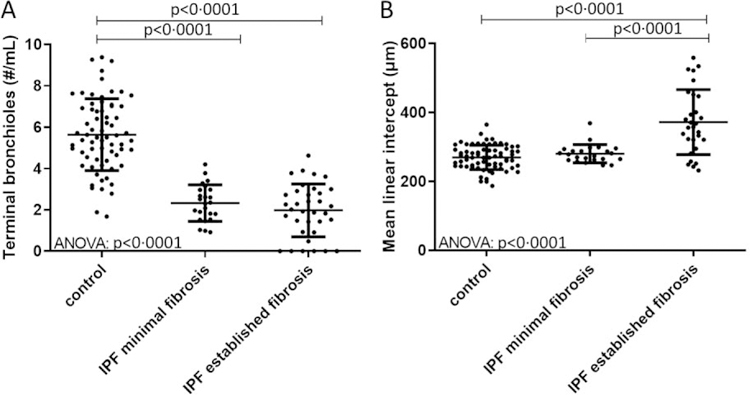
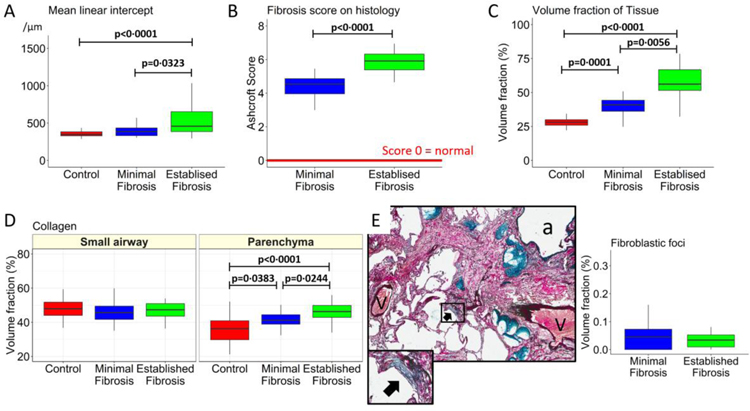
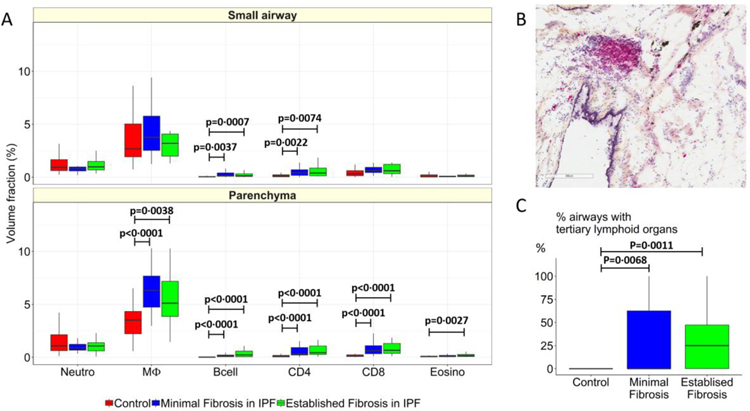
Findings: Between Oct 5, 2009, and July 22, 2016, explanted lungs from patients with severe IPF (n=11), were compared with a cohort of unused donor (control) lungs (n=10), providing 240 samples of lung tissue for comparison using the multiresolution imaging approach. The MDCT specimen scans show that the number of visible airways located between the ninth generation (control 69 [SD 22] versus patients with IPF 105 [33], p=0·0023) and 14th generation (control 9 [6] versus patients with IPF 49 [28], p<0·0001) of airway branching are increased in patients with IPF, which we show by micro-CT is due to thickening of their walls and distortion of their lumens. The micro-CT analysis showed that compared with healthy (control) lung anatomy (mean 5·6 terminal bronchioles per mL [SD 1·6]), minimal fibrosis in IPF tissue was associated with a 57% loss of the terminal bronchioles (mean 2·4 terminal bronchioles per mL [SD 1·0]; p<0·0001), the appearance of fibroblastic foci, and infiltration of the tissue by inflammatory immune cells capable of forming lymphoid follicles. Established fibrosis in IPF tissue had a similar reduction (66%) in the number of terminal bronchioles (mean 1·9 terminal bronchioles per mL [SD 1·4]; p<0·0001) and was dominated by increased airspace size, Ashcroft fibrosis score, and volume fractions of tissue and collagen.
Interpretation: Small airways disease is a feature of IPF, with significant loss of terminal bronchioles occuring within regions of minimal fibrosis. On the basis of these findings, we postulate that the small airways could become a potential therapeutic target in IPF.
Funding: Katholieke Universiteit Leuven, US National Institutes of Health, BC Lung Association, and Genentech.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
All other authors declare no competing interests.
Figures
Comment in
-
Dissecting the role of the small airways in idiopathic pulmonary fibrosis.Lancet Respir Med. 2020 Jun;8(6):529-531. doi: 10.1016/S2213-2600(20)30057-6. Epub 2020 Feb 13. Lancet Respir Med. 2020. PMID: 32061336 No abstract available.
References
-
- Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. American journal of respiratory and critical care medicine 2018; 198(5): e44–e68. - PubMed
-
- Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377(9779): 1760–9. - PubMed
-
- Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370(22): 2071–82. - PubMed
-
- Liebow AA CC. The interstitial pneumonias, frontiers of pulmonary radiology. New York: Grune and Stratton; 1969.
-
- Katzenstein AL, Mukhopadhyay S, Myers JL. Erratum to “Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases” [Hum Pathol 39 (2008) 1275–1294]. Hum Pathol 2008; 39(11): 1562–81. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
