

Mosaicism in Fanconi anemia: concise review and evaluation of published cases with focus on clinical course of blood count normalization
- PMID: 32065290
- PMCID: PMC7196946
- DOI: 10.1007/s00277-020-03954-2
Mosaicism in Fanconi anemia: concise review and evaluation of published cases with focus on clinical course of blood count normalization
Abstract
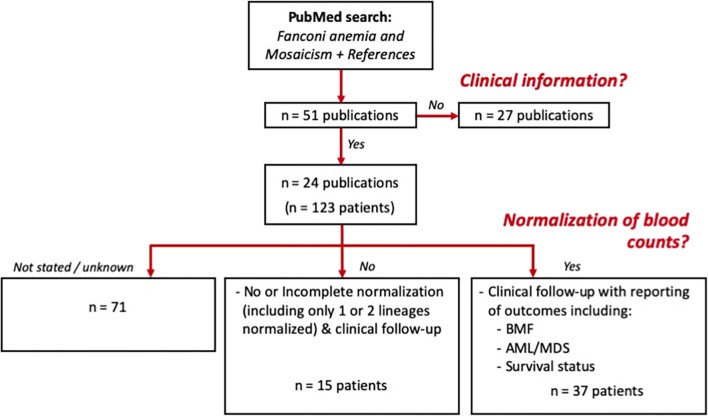
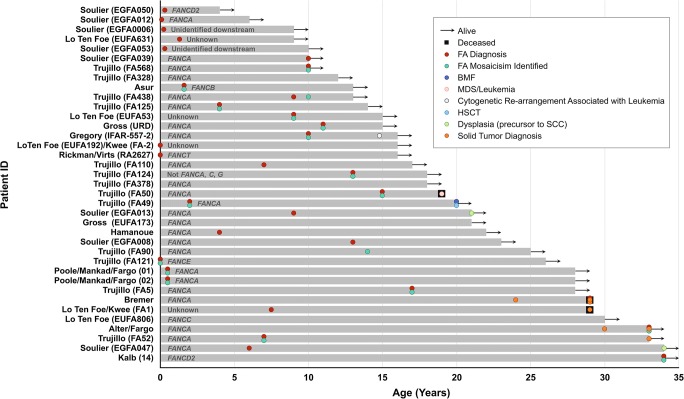
Fanconi anemia (FA) is a DNA repair disorder resulting from mutations in genes encoding for FA DNA repair complex components and is characterized by variable congenital abnormalities, bone marrow failure (BMF), and high incidences of malignancies. FA mosaicism arises from reversion or other compensatory mutations in hematopoietic cells and may be associated with BMF reversal and decreased blood cell sensitivity to DNA-damaging agents (clastogens); this sensitivity is a phenotypic and diagnostic hallmark of FA. Uncertainty regarding the clinical significance of FA mosaicism persists; in some cases, patients have survived multiple decades without BMF or hematologic malignancy, and in others hematologic failure occurred despite the presence of clastogen-resistant cell populations. Assessment of mosaicism is further complicated because clinical evaluation is frequently based on clastogen resistance in lymphocytes, which may arise from reversion events both in lymphoid-specific lineages and in more pluripotent hematopoietic stem/progenitor cells (HSPCs). In this review, we describe diagnostic methods and outcomes in published mosaicism series, including the substantial intervals (1-6 years) over which blood counts normalized, and the relatively favorable clinical course in cases where clastogen resistance was demonstrated in bone marrow progenitors. We also analyzed published FA mosaic cases with emphasis on long-term clinical outcomes when blood count normalization was identified. Blood count normalization in FA mosaicism likely arises from reversion events in long-term primitive HSPCs and is associated with low incidences of BMF or hematologic malignancy. These observations have ramifications for current investigational therapeutic programs in FA intended to enable gene correction in long-term repopulating HSPCs.
Keywords: Autologous stem cell transplantation; Bone marrow failure; Fanconi anemia; Gene therapy; Mosaicism.
Conflict of interest statement
Eileen Nicoletti is an employee of Rocket Pharmaceuticals, Inc. Gayatri Rao is an employee of Rocket Pharmaceuticals, Inc. Juan A. Bueren is the head of the Hematopoietic Innovative Therapies Division at Centro de Investigaciones Energeticas, Medioambientales y Tecnologicas (CIEMAT) which receives funding for Fanconi Anemia (FA) gene therapy and has licensed the PGK-FANCA-Wpre* lentiviral vector to Rocket Pharmaceuticals, Inc. He is also a consultant for Rocket Pharmaceuticals, Inc. Paula Río is a member of the Hematopoietic Innovative Therapies Division at Centro de Investigaciones Energeticas, Medioambientales y Tecnologicas (CIEMAT) which receives funding for Fanconi Anemia (FA) gene therapy and has licensed the PGK-FANCA-Wpre* lentiviral vector to Rocket Pharmaceuticals, Inc. Susana Navarro is a member of the Hematopoietic Innovative Therapies Division at Centro de Investigaciones Energeticas, Medioambientales y Tecnologicas (CIEMAT) which receives funding for Fanconi Anemia (FA) gene therapy and has licensed the PGK-FANCA-Wpre* lentiviral vector to Rocket Pharmaceuticals, Inc. Jordi Surrallés does not have any conflicts of interest. Grace Choi is an employee of Rocket Pharmaceuticals, Inc. Jonathan D. Schwartz is an employee of Rocket Pharmaceuticals, Inc.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
