

Dominant mutations of the Notch ligand Jagged1 cause peripheral neuropathy
- PMID: 32065591
- PMCID: PMC7269582
- DOI: 10.1172/JCI128152
Dominant mutations of the Notch ligand Jagged1 cause peripheral neuropathy
Abstract
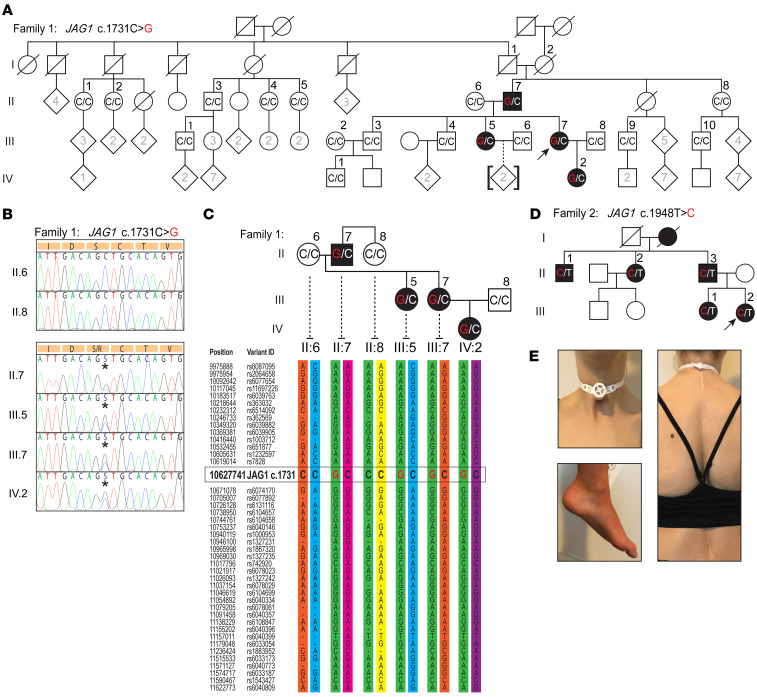
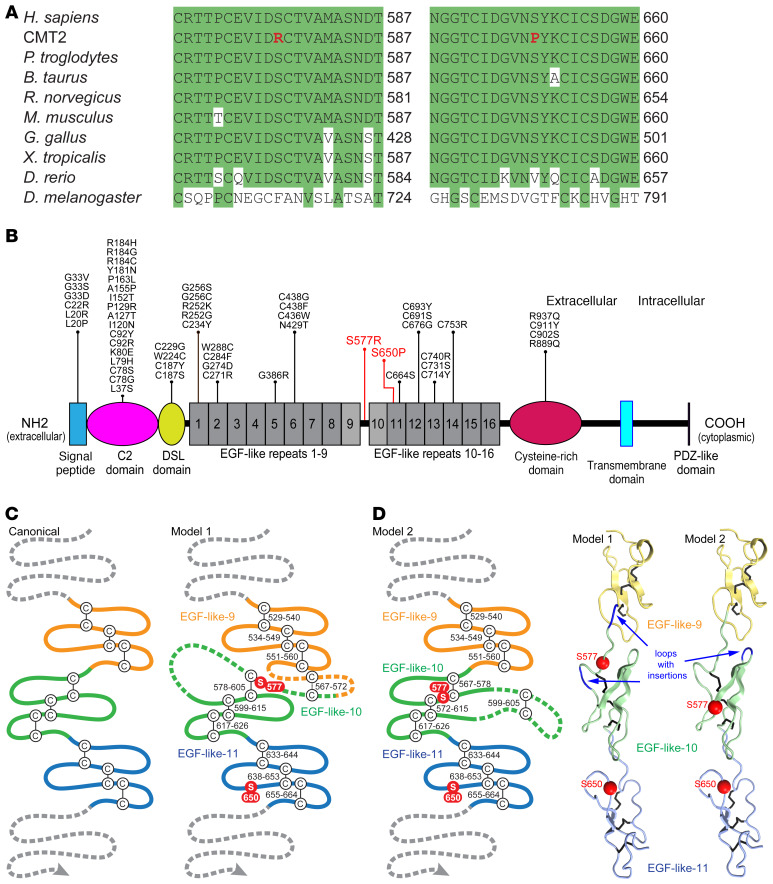
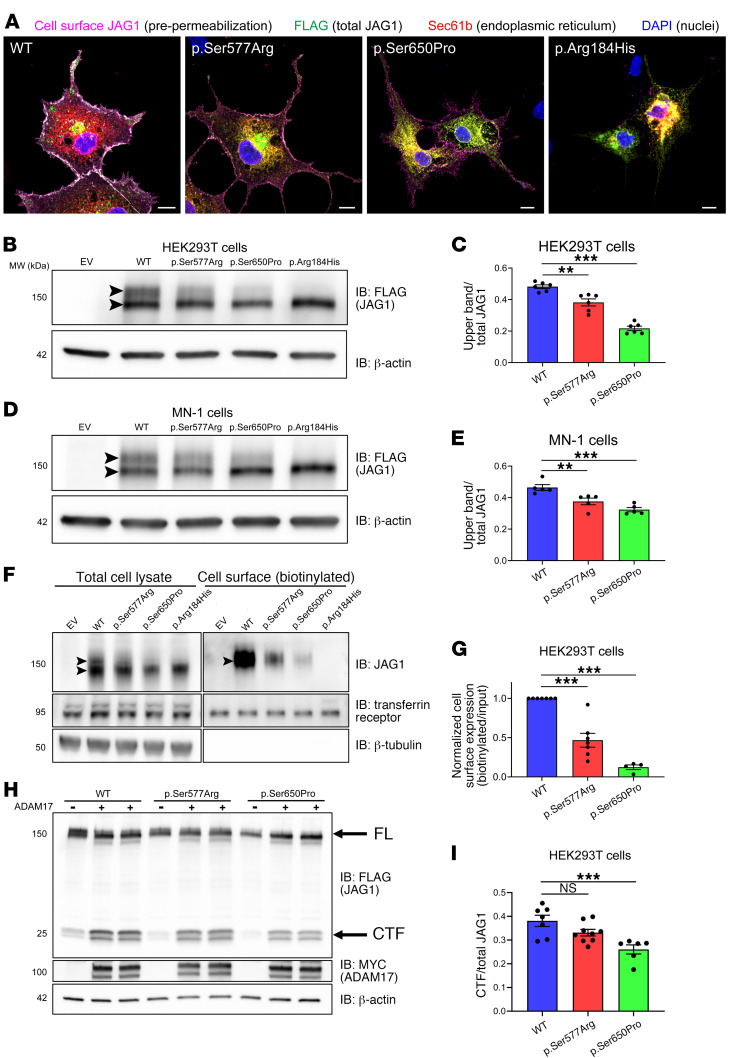
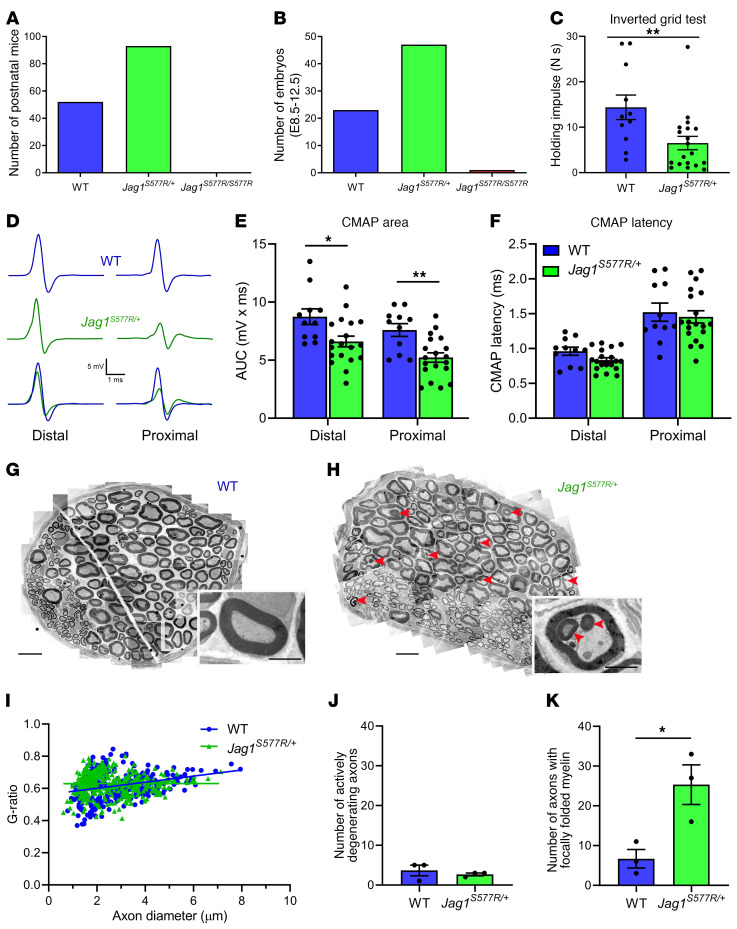
Notch signaling is a highly conserved intercellular pathway with tightly regulated and pleiotropic roles in normal tissue development and homeostasis. Dysregulated Notch signaling has also been implicated in human disease, including multiple forms of cancer, and represents an emerging therapeutic target. Successful development of such therapeutics requires a detailed understanding of potential on-target toxicities. Here, we identify autosomal dominant mutations of the canonical Notch ligand Jagged1 (or JAG1) as a cause of peripheral nerve disease in 2 unrelated families with the hereditary axonal neuropathy Charcot-Marie-Tooth disease type 2 (CMT2). Affected individuals in both families exhibited severe vocal fold paresis, a rare feature of peripheral nerve disease that can be life-threatening. Our studies of mutant protein posttranslational modification and localization indicated that the mutations (p.Ser577Arg, p.Ser650Pro) impair protein glycosylation and reduce JAG1 cell surface expression. Mice harboring heterozygous CMT2-associated mutations exhibited mild peripheral neuropathy, and homozygous expression resulted in embryonic lethality by midgestation. Together, our findings highlight a critical role for JAG1 in maintaining peripheral nerve integrity, particularly in the recurrent laryngeal nerve, and provide a basis for the evaluation of peripheral neuropathy as part of the clinical development of Notch pathway-modulating therapeutics.
Keywords: Genetic diseases; Genetics; Neurodegeneration; Neuromuscular disease; Neuroscience.
Conflict of interest statement
Figures
References
-
- Braune EB, Lendahl U. Notch -- a Goldilocks signaling pathway in disease and cancer therapy. Discov Med. 2016;21(115):189–196. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
