

A census of pathway maps in cancer systems biology
- PMID: 32066900
- PMCID: PMC7224610
- DOI: 10.1038/s41568-020-0240-7
A census of pathway maps in cancer systems biology
Erratum in
-
Author Correction: A census of pathway maps in cancer systems biology.Nat Rev Cancer. 2021 Mar;21(3):212. doi: 10.1038/s41568-021-00331-7. Nat Rev Cancer. 2021. PMID: 33441978 No abstract available.
Abstract
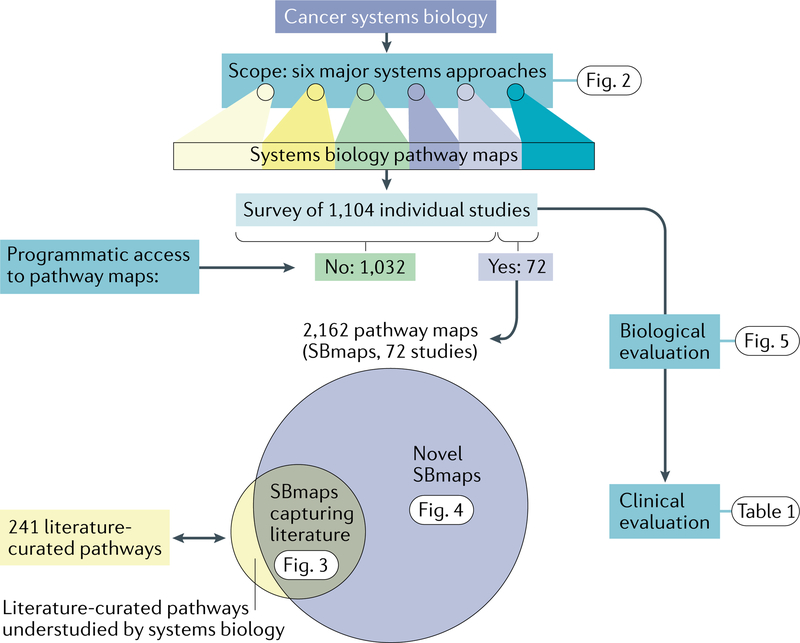
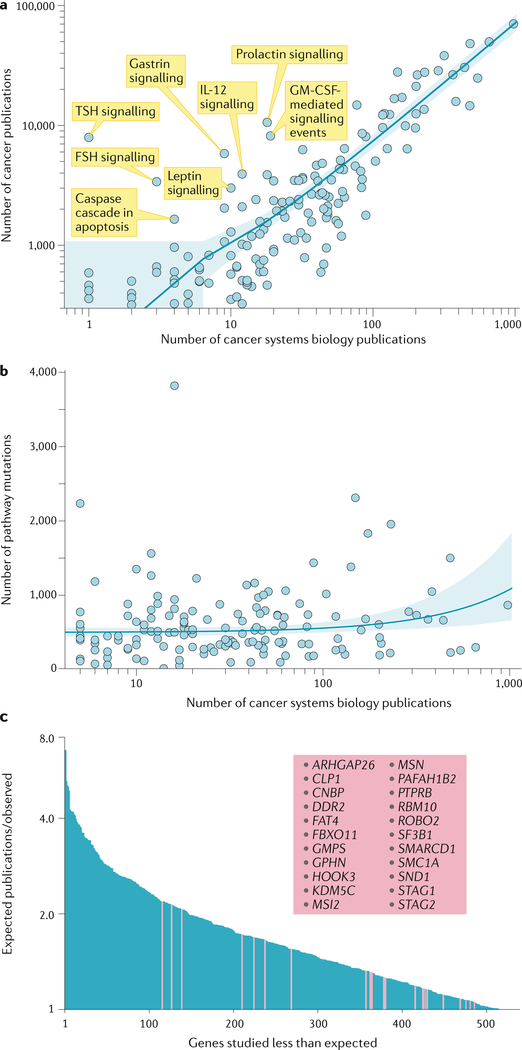
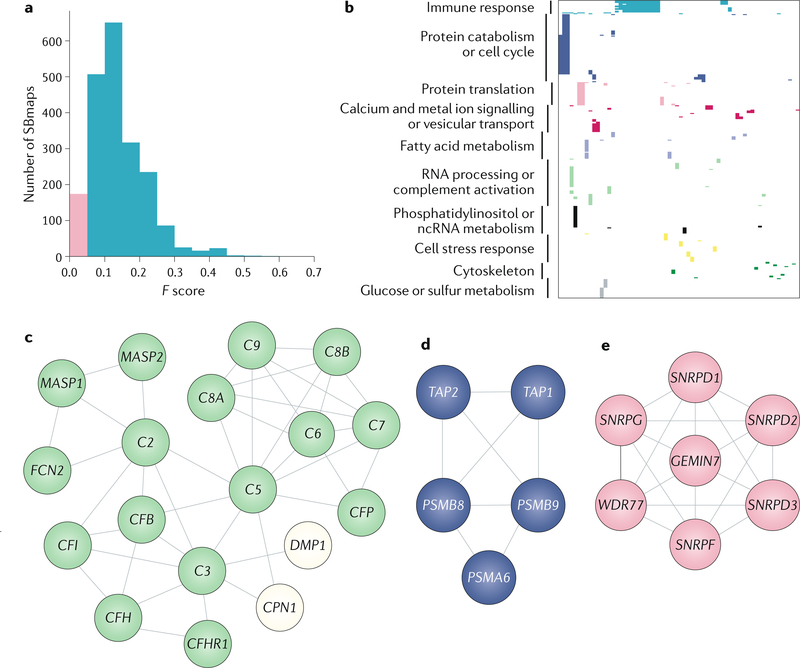
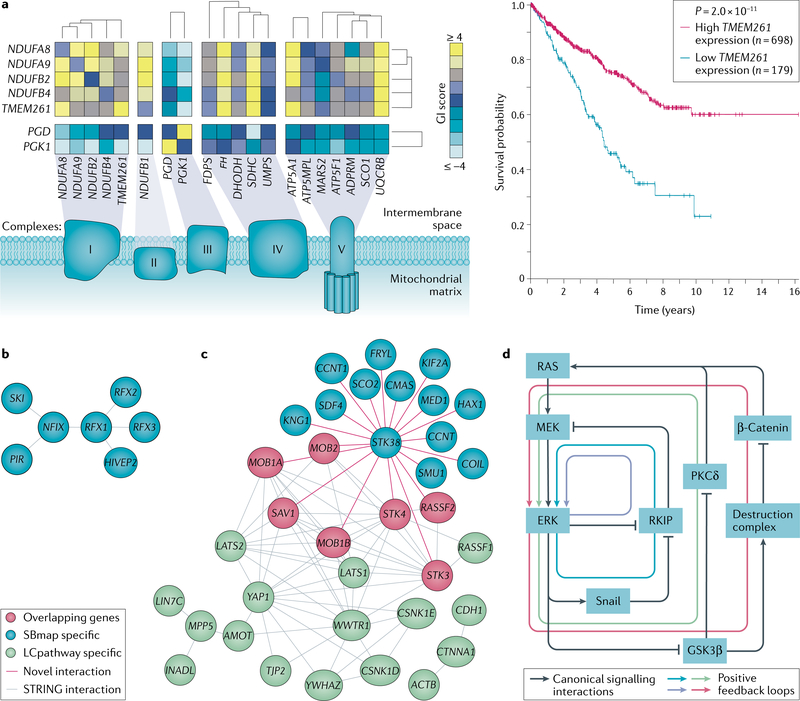
A key goal of cancer systems biology is to use big data to elucidate the molecular networks by which cancer develops. However, to date there has been no systematic evaluation of how far these efforts have progressed. In this Analysis, we survey six major systems biology approaches for mapping and modelling cancer pathways with attention to how well their resulting network maps cover and enhance current knowledge. Our sample of 2,070 systems biology maps captures all literature-curated cancer pathways with significant enrichment, although the strong tendency is for these maps to recover isolated mechanisms rather than entire integrated processes. Systems biology maps also identify previously underappreciated functions, such as a potential role for human papillomavirus-induced chromosomal alterations in ovarian tumorigenesis, and they add new genes to known cancer pathways, such as those related to metabolism, Hippo signalling and immunity. Notably, we find that many cancer networks have been provided only in journal figures and not for programmatic access, underscoring the need to deposit network maps in community databases to ensure they can be readily accessed. Finally, few of these findings have yet been clinically translated, leaving ample opportunity for future translational studies. Periodic surveys of cancer pathway maps, such as the one reported here, are critical to assess progress in the field and identify underserved areas of methodology and cancer biology.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures


Similar articles
-
Metabolic and signalling network maps integration: application to cross-talk studies and omics data analysis in cancer.BMC Bioinformatics. 2019 Apr 18;20(Suppl 4):140. doi: 10.1186/s12859-019-2682-z. BMC Bioinformatics. 2019. PMID: 30999838 Free PMC article.
-
Comparative analysis of protein interactome networks prioritizes candidate genes with cancer signatures.Oncotarget. 2016 Nov 29;7(48):78841-78849. doi: 10.18632/oncotarget.12879. Oncotarget. 2016. PMID: 27791983 Free PMC article.
-
Introduction: Cancer Gene Networks.Methods Mol Biol. 2017;1513:1-9. doi: 10.1007/978-1-4939-6539-7_1. Methods Mol Biol. 2017. PMID: 27807826
-
Decoding and targeting the molecular basis of MACC1-driven metastatic spread: Lessons from big data mining and clinical-experimental approaches.Semin Cancer Biol. 2020 Feb;60:365-379. doi: 10.1016/j.semcancer.2019.08.010. Epub 2019 Aug 17. Semin Cancer Biol. 2020. PMID: 31430556 Review.
-
Community-driven roadmap for integrated disease maps.Brief Bioinform. 2019 Mar 25;20(2):659-670. doi: 10.1093/bib/bby024. Brief Bioinform. 2019. PMID: 29688273 Free PMC article. Review.
Cited by
-
Learning from cancer to address COVID-19.Biol Futur. 2023 Jun;74(1-2):29-43. doi: 10.1007/s42977-023-00156-5. Epub 2023 Jul 6. Biol Futur. 2023. PMID: 37410273 Review.
-
Cystatin SN promotes epithelial-mesenchymal transition and serves as a prognostic biomarker in lung adenocarcinoma.BMC Cancer. 2022 May 30;22(1):589. doi: 10.1186/s12885-022-09685-z. BMC Cancer. 2022. PMID: 35637432 Free PMC article.
-
CancerVar: An artificial intelligence-empowered platform for clinical interpretation of somatic mutations in cancer.Sci Adv. 2022 May 6;8(18):eabj1624. doi: 10.1126/sciadv.abj1624. Epub 2022 May 6. Sci Adv. 2022. PMID: 35544644 Free PMC article.
-
Co-targeting FAK and Gli1 inhibits the tumor-associated macrophages-released CCL22-mediated esophageal squamous cell carcinoma malignancy.MedComm (2020). 2023 Oct 15;4(6):e381. doi: 10.1002/mco2.381. eCollection 2023 Dec. MedComm (2020). 2023. PMID: 37846367 Free PMC article.
-
YAP1 is a Prognostic Biomarker and Correlated with Immune Cell Infiltration in Pancreatic Cancer.Front Mol Biosci. 2021 Jun 2;8:625731. doi: 10.3389/fmolb.2021.625731. eCollection 2021. Front Mol Biosci. 2021. PMID: 34150844 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
