

An immune-cell signature of bacterial sepsis
- PMID: 32066974
- PMCID: PMC7235950
- DOI: 10.1038/s41591-020-0752-4
An immune-cell signature of bacterial sepsis
Abstract
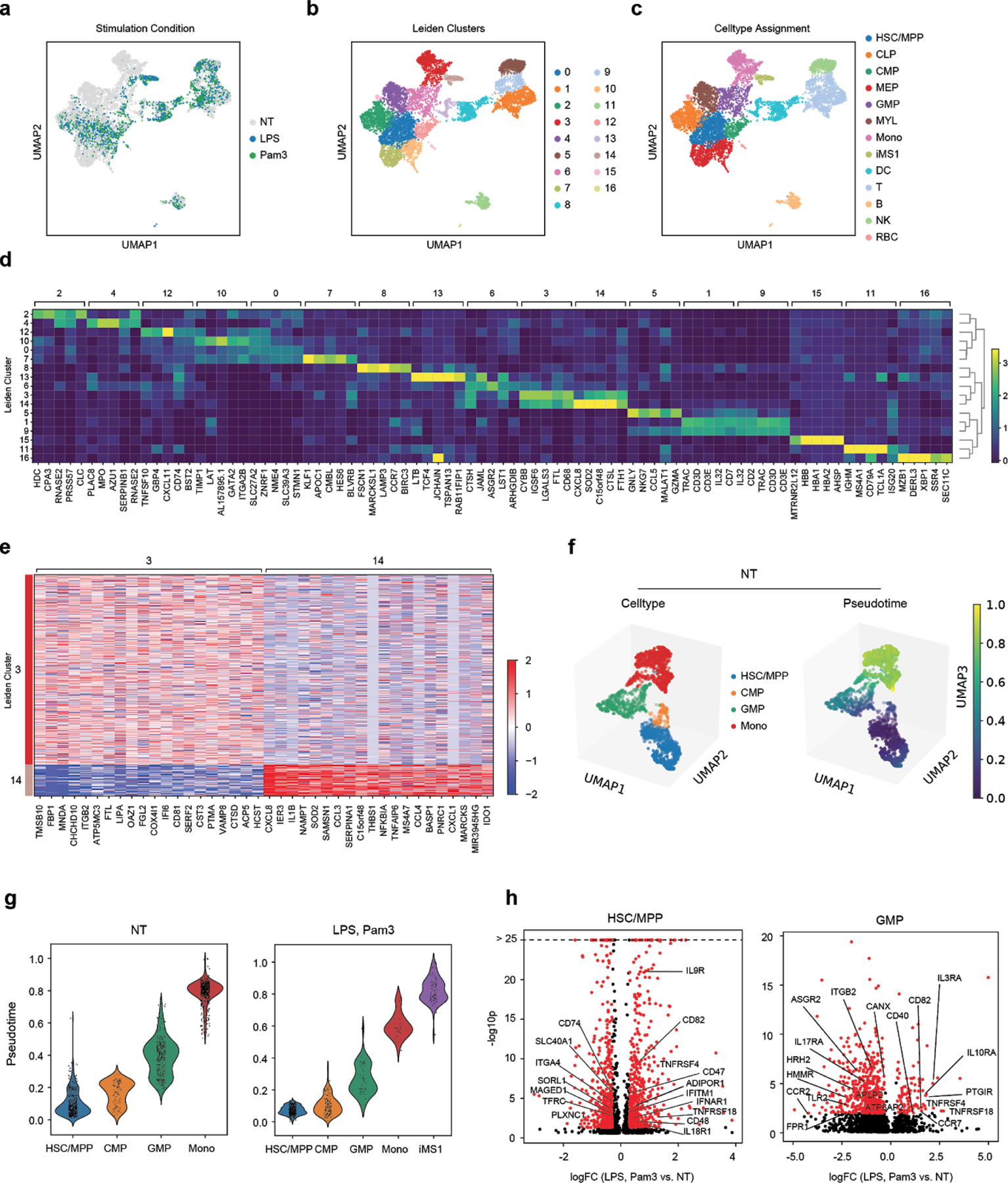
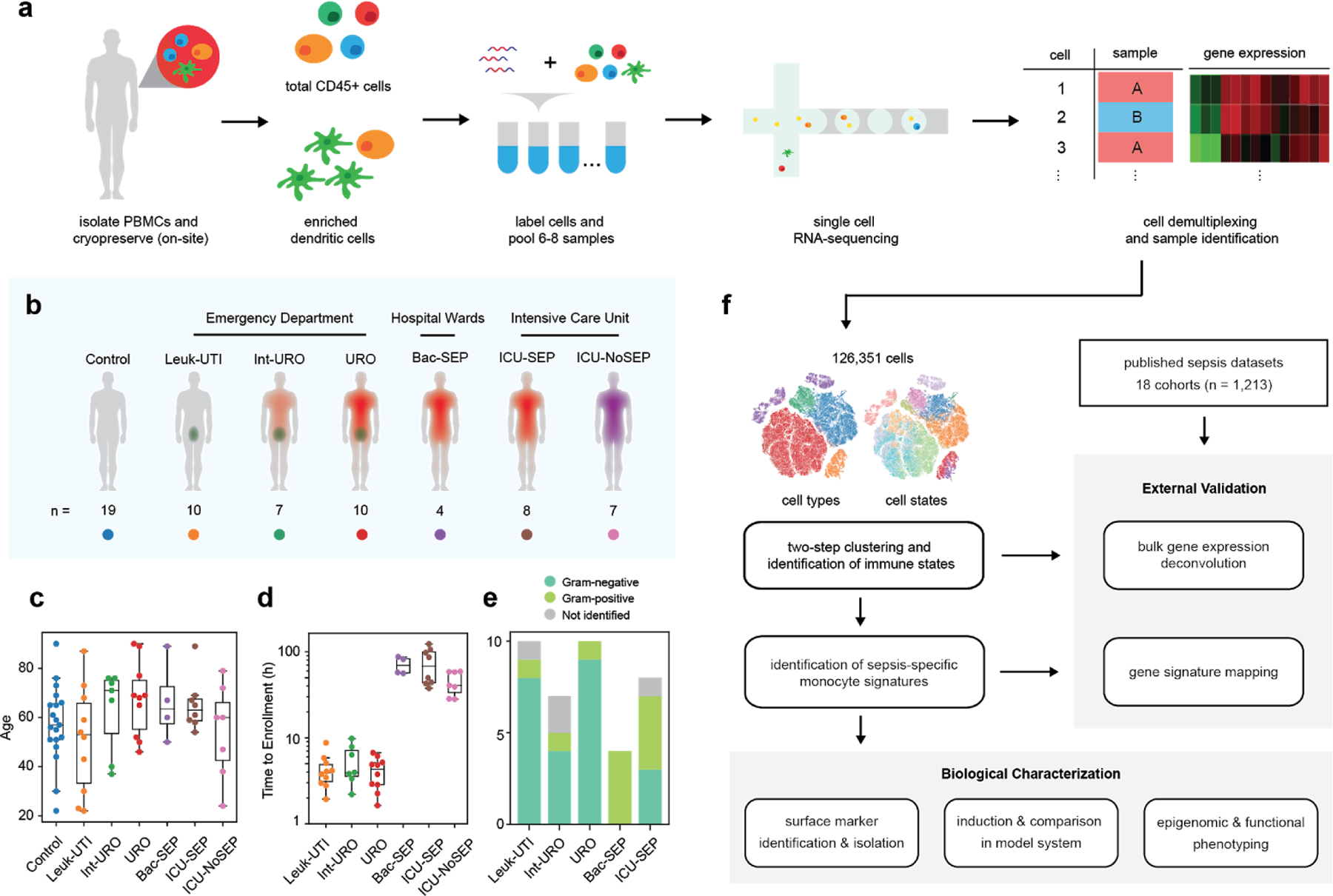
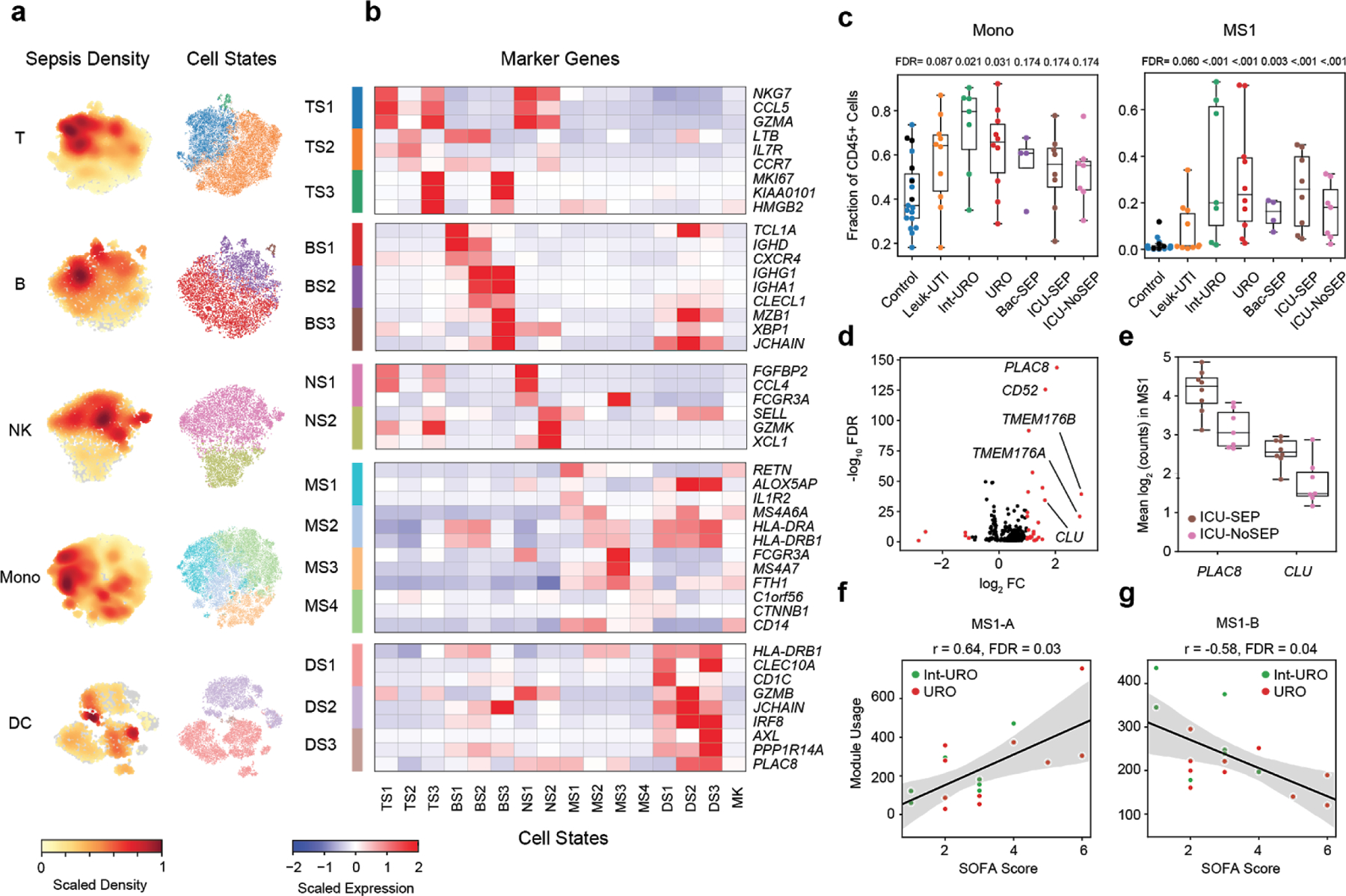
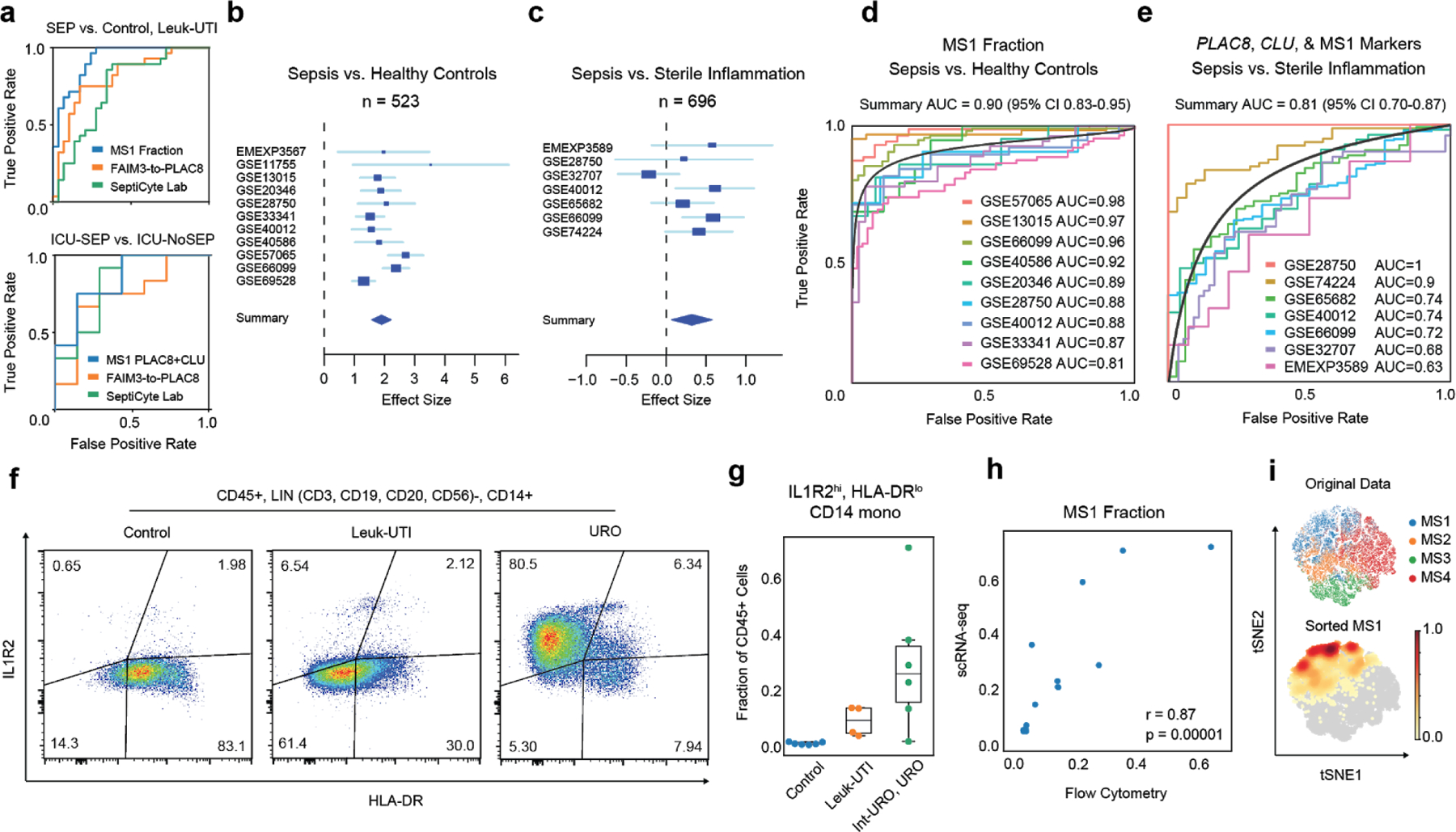
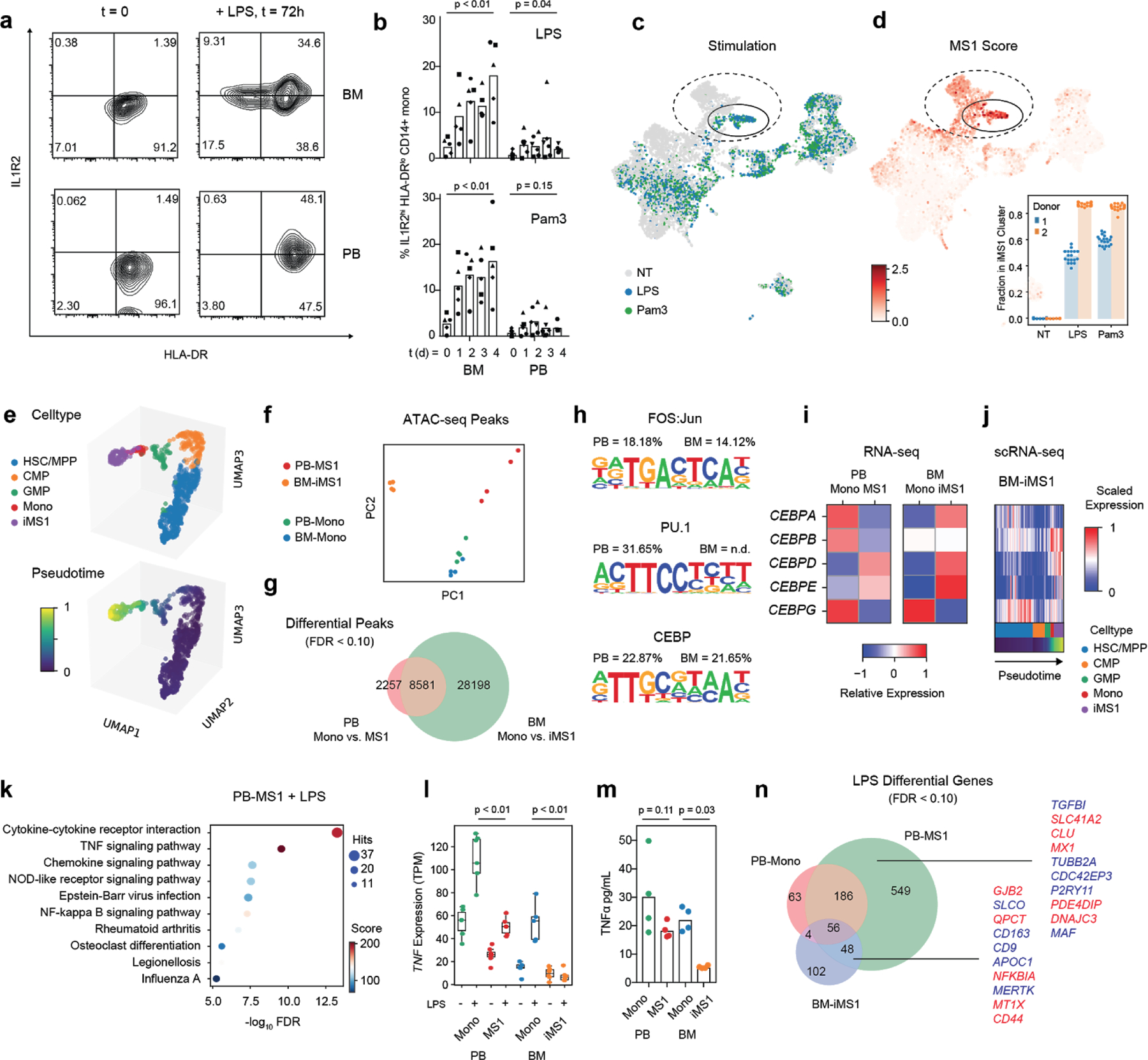
Dysregulation of the immune response to bacterial infection can lead to sepsis, a condition with high mortality. Multiple whole-blood gene-expression studies have defined sepsis-associated molecular signatures, but have not resolved changes in transcriptional states of specific cell types. Here, we used single-cell RNA-sequencing to profile the blood of people with sepsis (n = 29) across three clinical cohorts with corresponding controls (n = 36). We profiled total peripheral blood mononuclear cells (PBMCs, 106,545 cells) and dendritic cells (19,806 cells) across all subjects and, on the basis of clustering of their gene-expression profiles, defined 16 immune-cell states. We identified a unique CD14+ monocyte state that is expanded in people with sepsis and validated its power in distinguishing these individuals from controls using public transcriptomic data from subjects with different disease etiologies and from multiple geographic locations (18 cohorts, n = 1,467 subjects). We identified a panel of surface markers for isolation and quantification of the monocyte state and characterized its epigenomic and functional phenotypes, and propose a model for its induction from human bone marrow. This study demonstrates the utility of single-cell genomics in discovering disease-associated cytologic signatures and provides insight into the cellular basis of immune dysregulation in bacterial sepsis.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
The Broad Institute and MIT may seek to commercialize aspects of this work, and related applications for intellectual property have been filed. In addition, P.C.B. is a consultant to and equity holder in a company, 10X Genomics, whose products were used in this study.
Figures
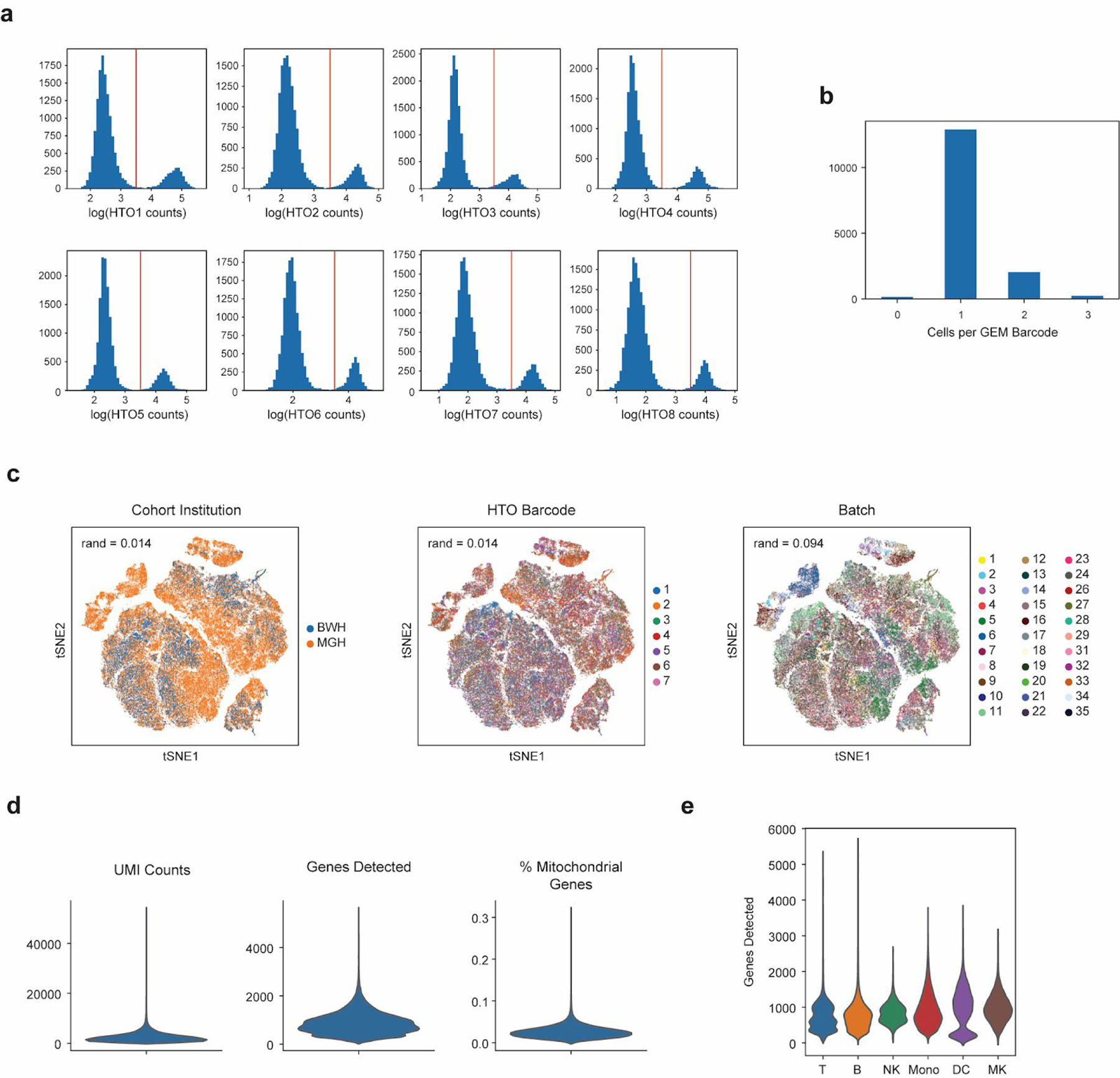
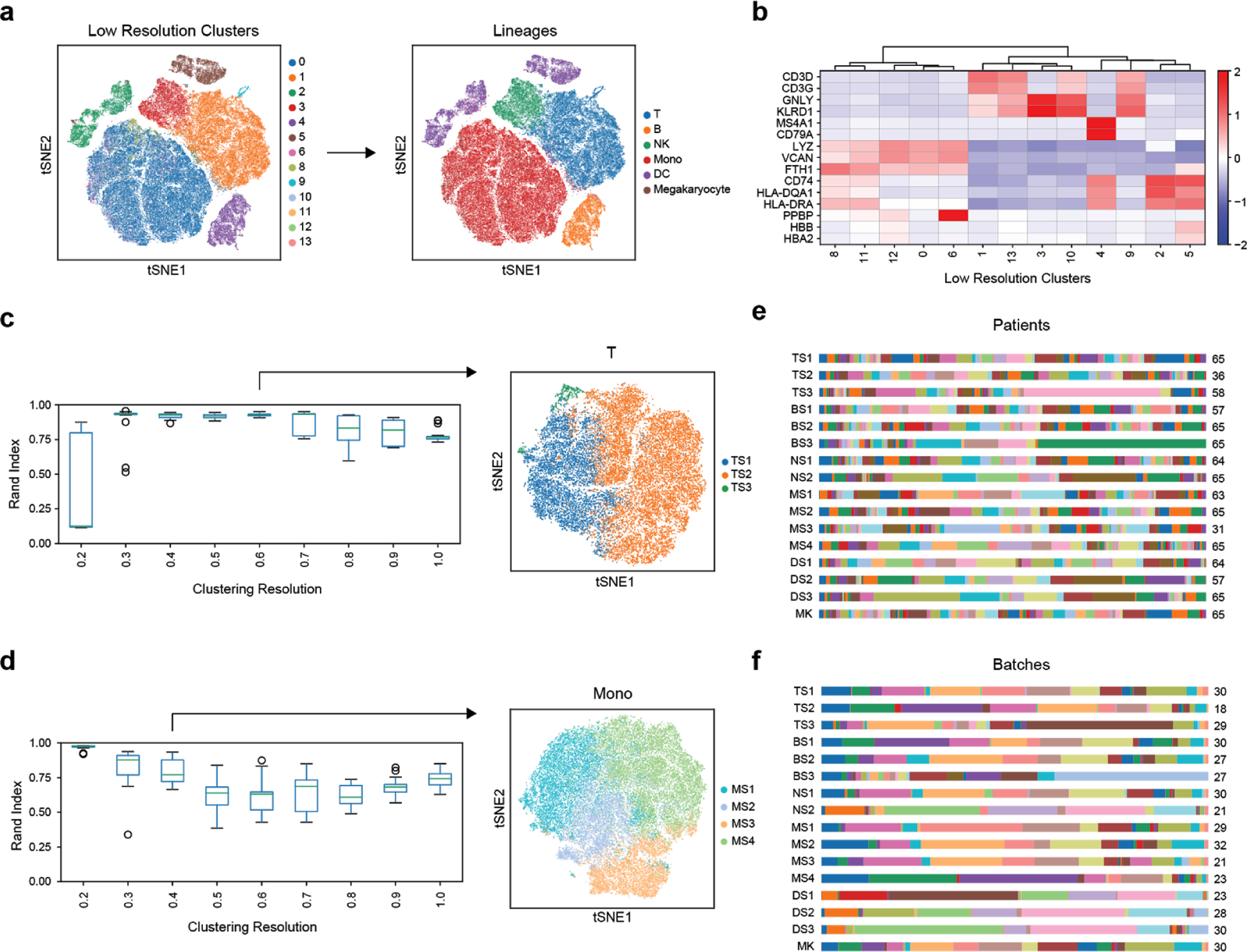
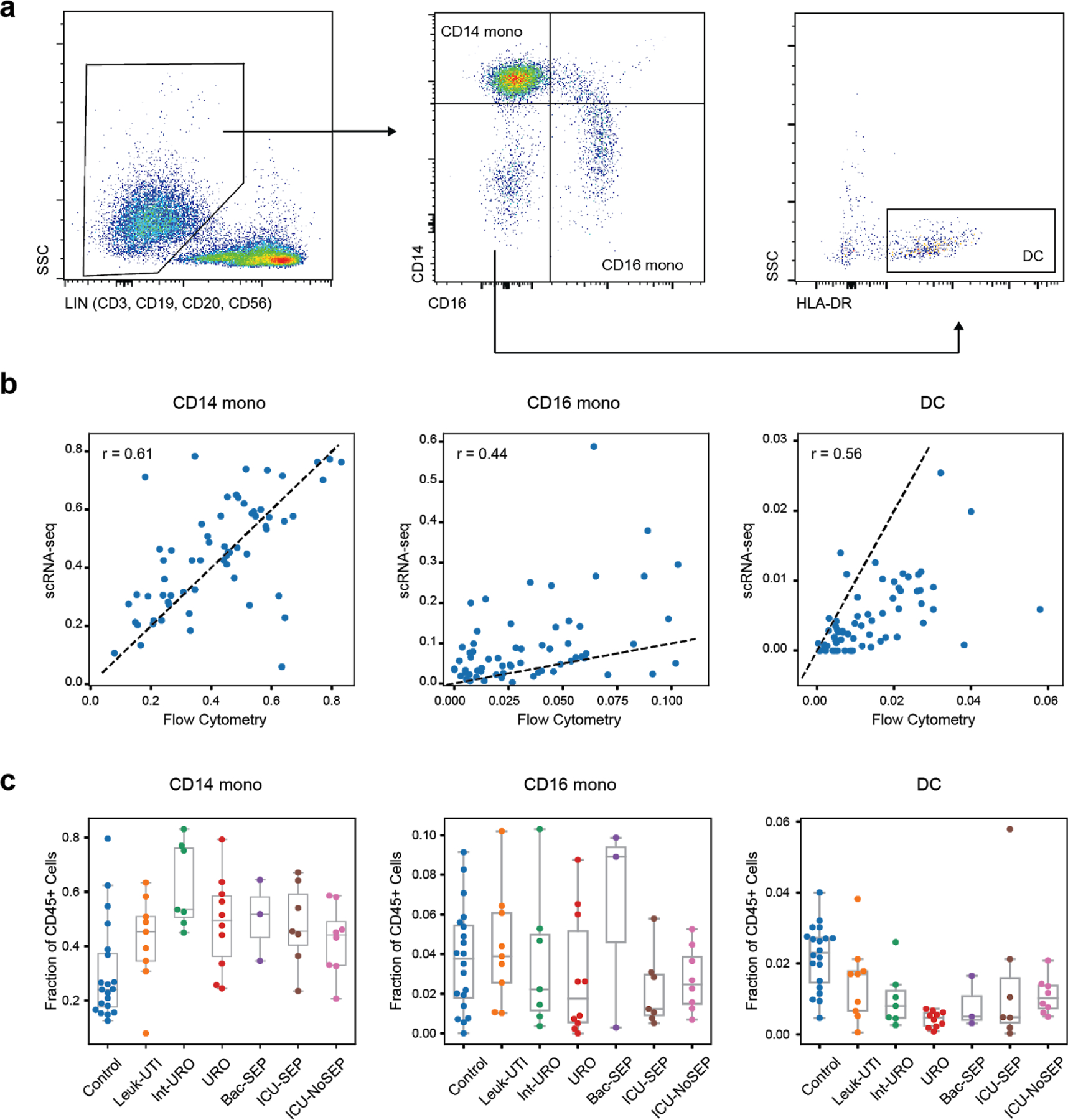
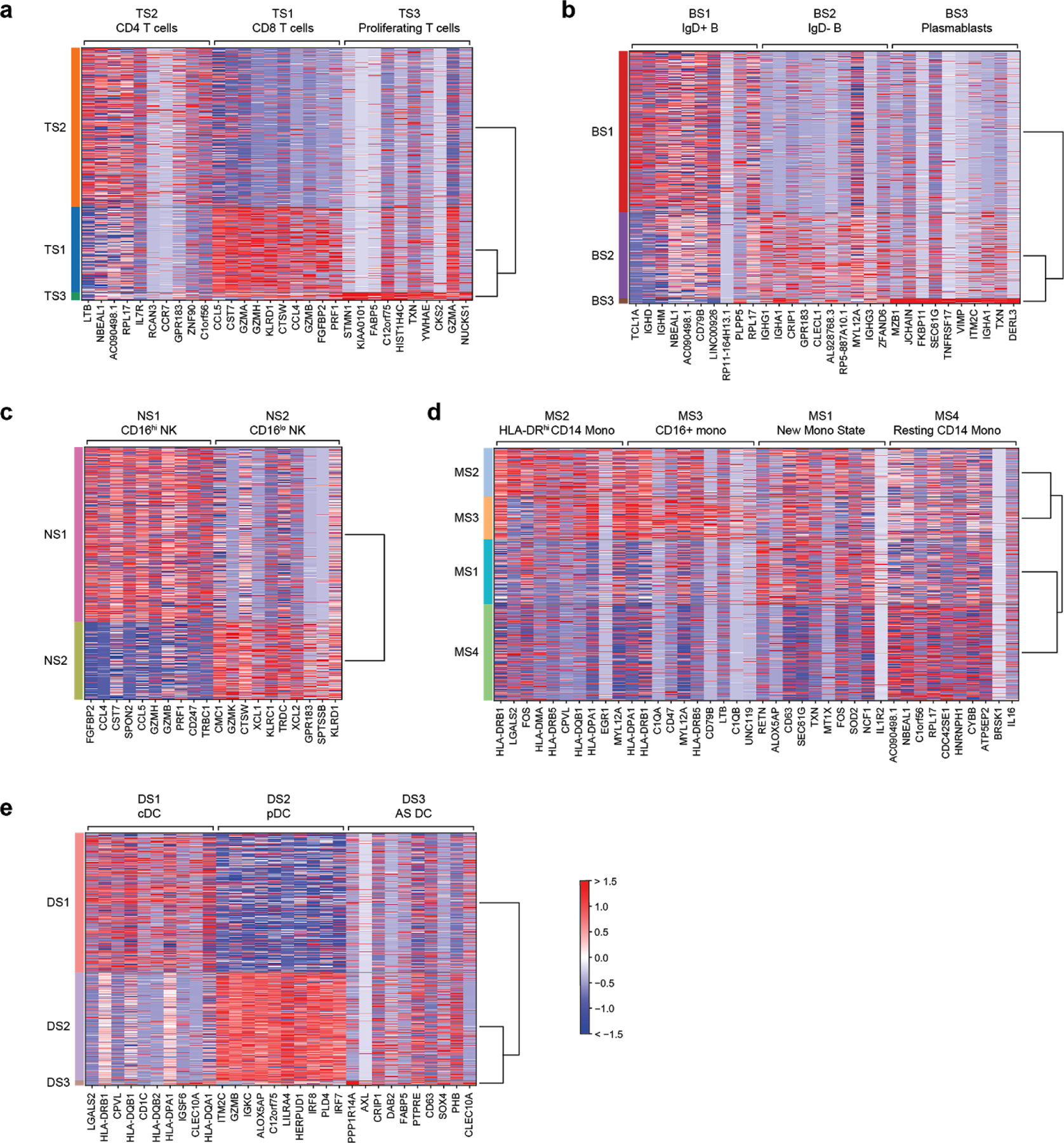
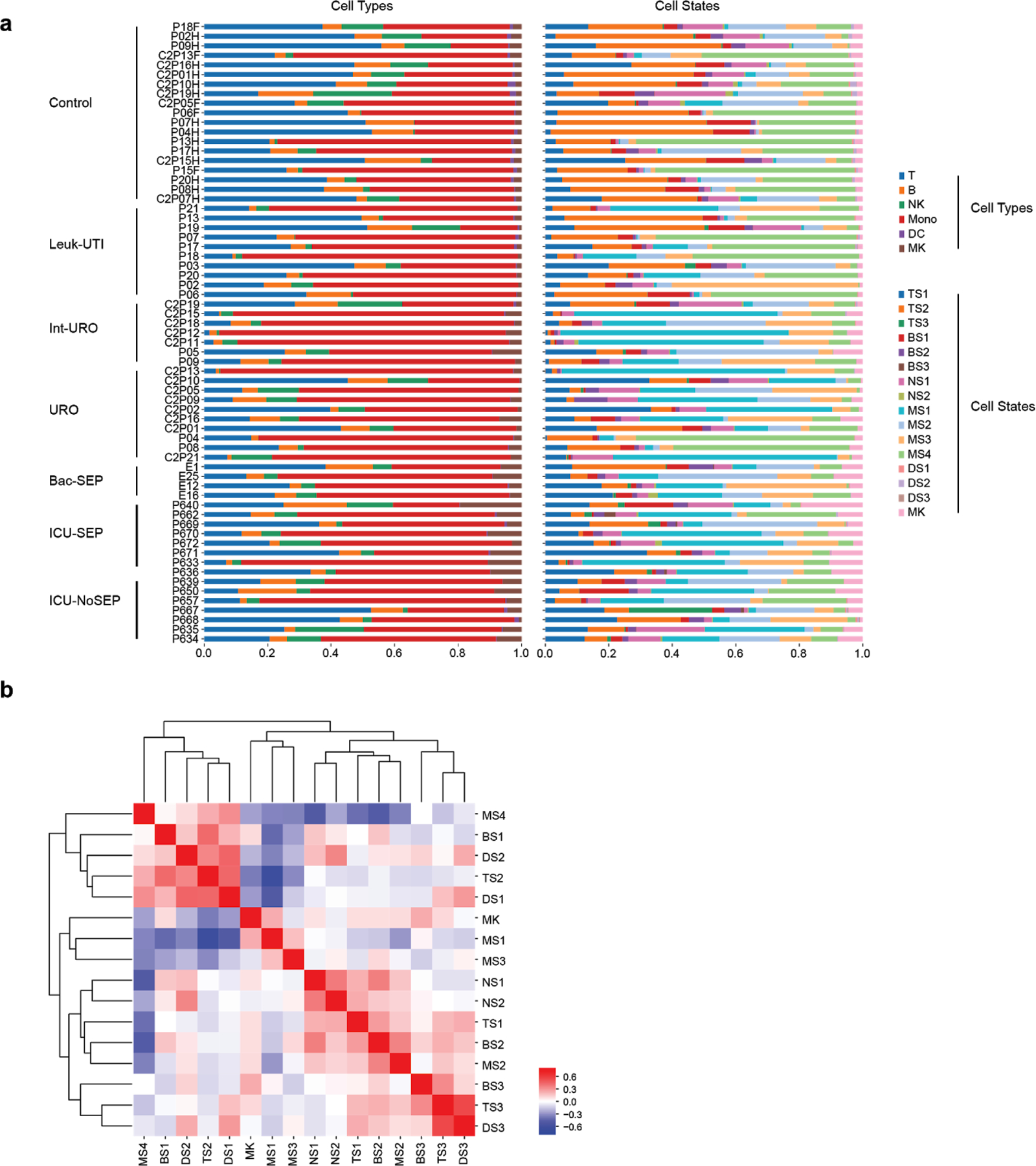

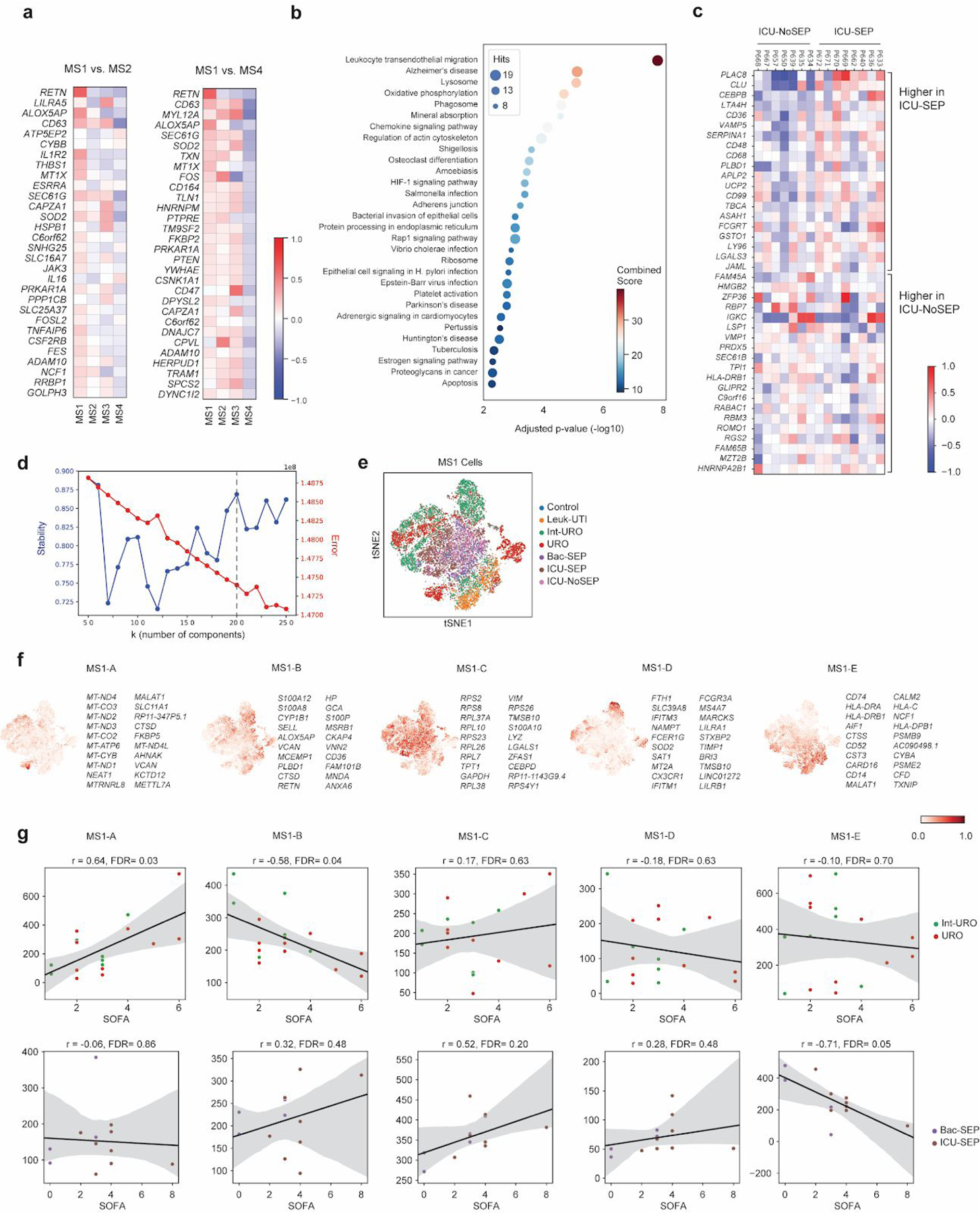
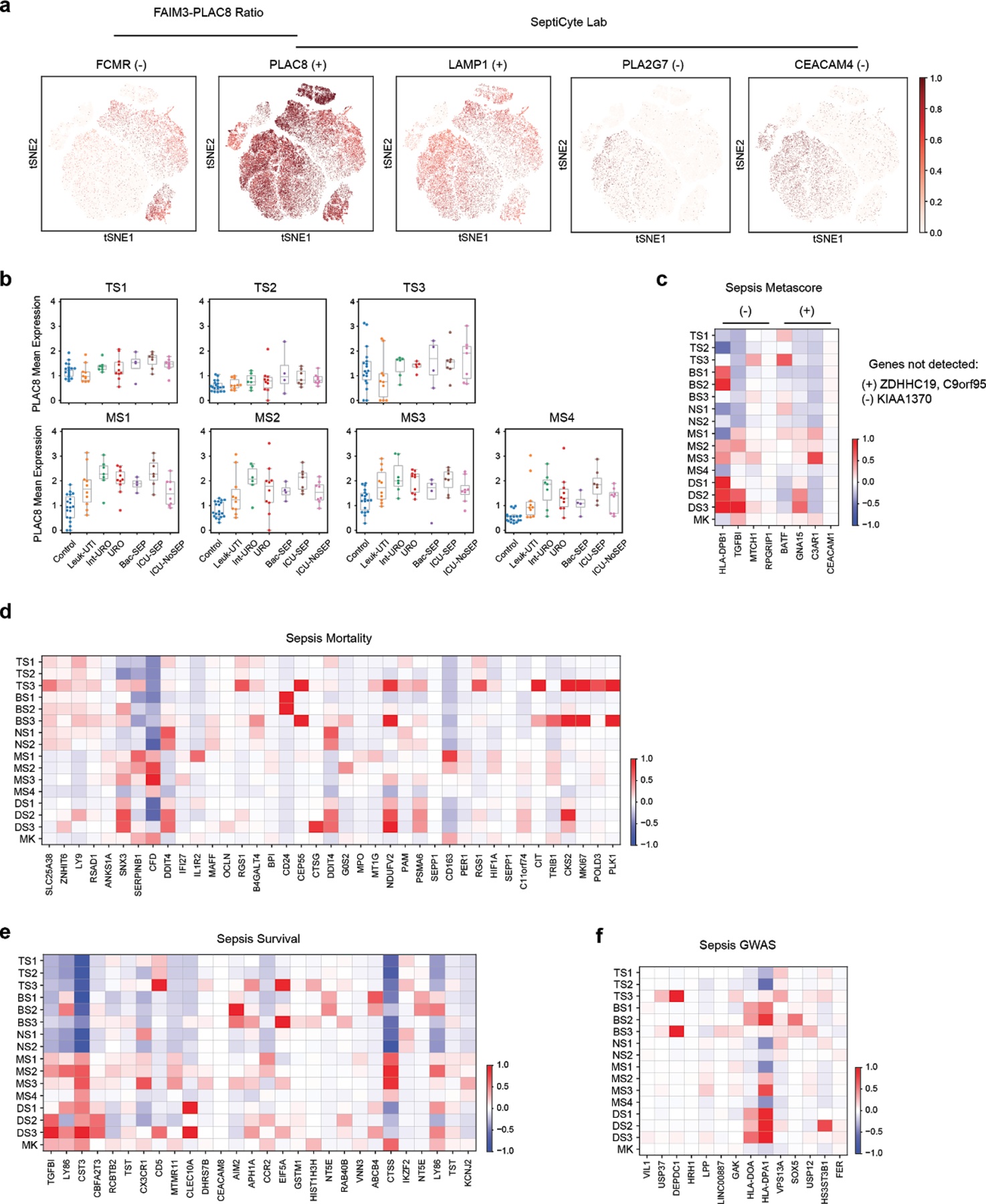
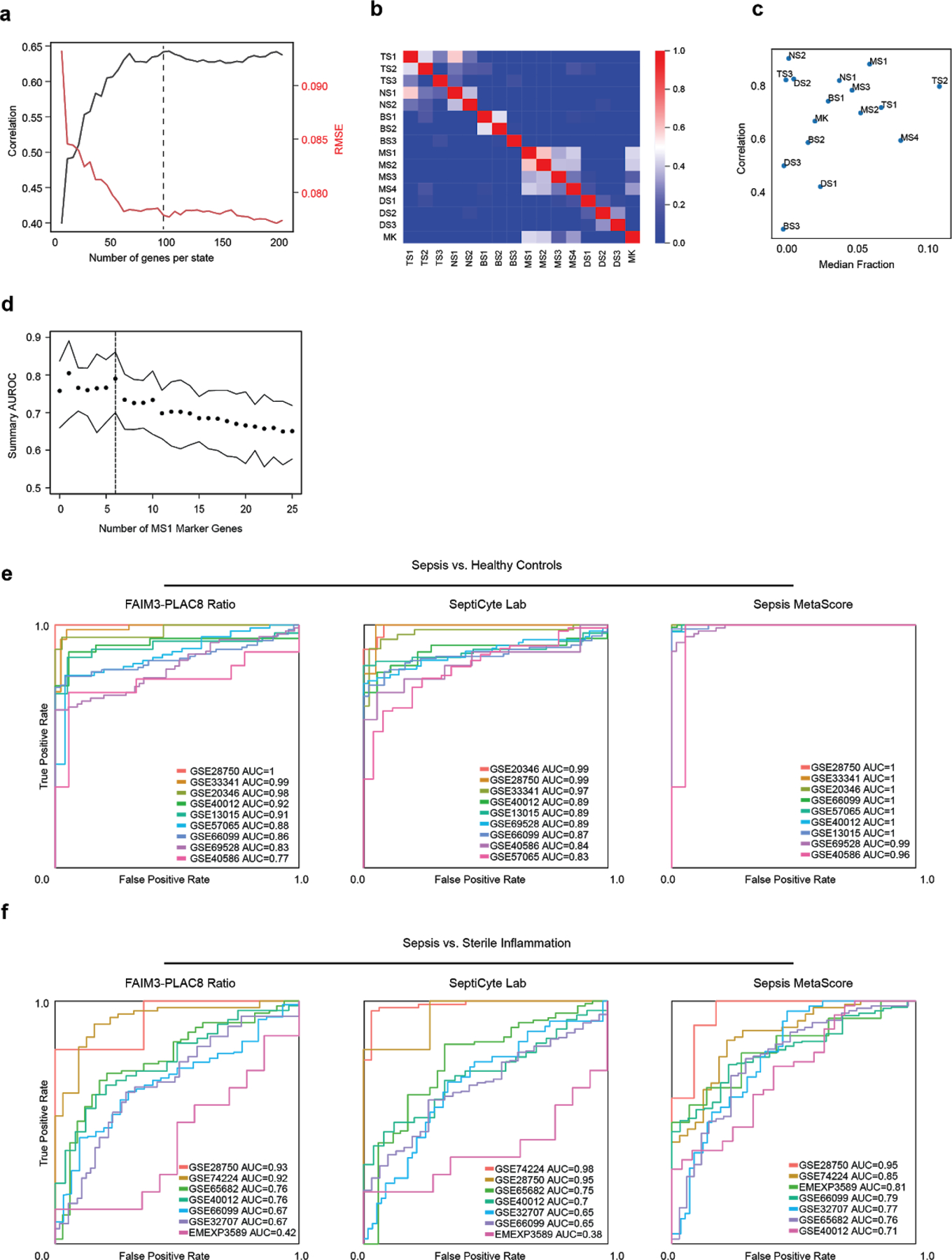
References
-
- Filbin MR et al. Presenting Symptoms Independently Predict Mortality in Septic Shock: Importance of a Previously Unmeasured Confounder. Crit. Care Med 46, 1592–1599 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL142093/HL/NHLBI NIH HHS/United States
- R56 AI137244/AI/NIAID NIH HHS/United States
- P50 HG006193/HG/NHGRI NIH HHS/United States
- U24 AI118668/AI/NIAID NIH HHS/United States
- U24AI118668/U.S. Department of Health & Human Services | NIH | National Institute of Allergy and Infectious Diseases (NIAID)/International
- K08 AI119157/AI/NIAID NIH HHS/United States
- 1DSEP160030-01-00/U.S. Department of Health & Human Services | Biomedical Advanced Research and Development Authority (BARDA)/International
- 1K08AI119157-04/U.S. Department of Health & Human Services | National Institutes of Health (NIH)/International
- RM1 HG006193/HG/NHGRI NIH HHS/United States
- U24 AI118656/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
