

BlobToolKit - Interactive Quality Assessment of Genome Assemblies
- PMID: 32071071
- PMCID: PMC7144090
- DOI: 10.1534/g3.119.400908
BlobToolKit - Interactive Quality Assessment of Genome Assemblies
Abstract
Reconstruction of target genomes from sequence data produced by instruments that are agnostic as to the species-of-origin may be confounded by contaminant DNA. Whether introduced during sample processing or through co-extraction alongside the target DNA, if insufficient care is taken during the assembly process, the final assembled genome may be a mixture of data from several species. Such assemblies can confound sequence-based biological inference and, when deposited in public databases, may be included in downstream analyses by users unaware of underlying problems. We present BlobToolKit, a software suite to aid researchers in identifying and isolating non-target data in draft and publicly available genome assemblies. BlobToolKit can be used to process assembly, read and analysis files for fully reproducible interactive exploration in the browser-based Viewer. BlobToolKit can be used during assembly to filter non-target DNA, helping researchers produce assemblies with high biological credibility. We have been running an automated BlobToolKit pipeline on eukaryotic assemblies publicly available in the International Nucleotide Sequence Data Collaboration and are making the results available through a public instance of the Viewer at https://blobtoolkit.genomehubs.org/view We aim to complete analysis of all publicly available genomes and then maintain currency with the flow of new genomes. We have worked to embed these views into the presentation of genome assemblies at the European Nucleotide Archive, providing an indication of assembly quality alongside the public record with links out to allow full exploration in the Viewer.
Keywords: Bioinformatics; genome assembly; quality control; visualisation web-tool.
Copyright © 2020 Challis et al.
Figures

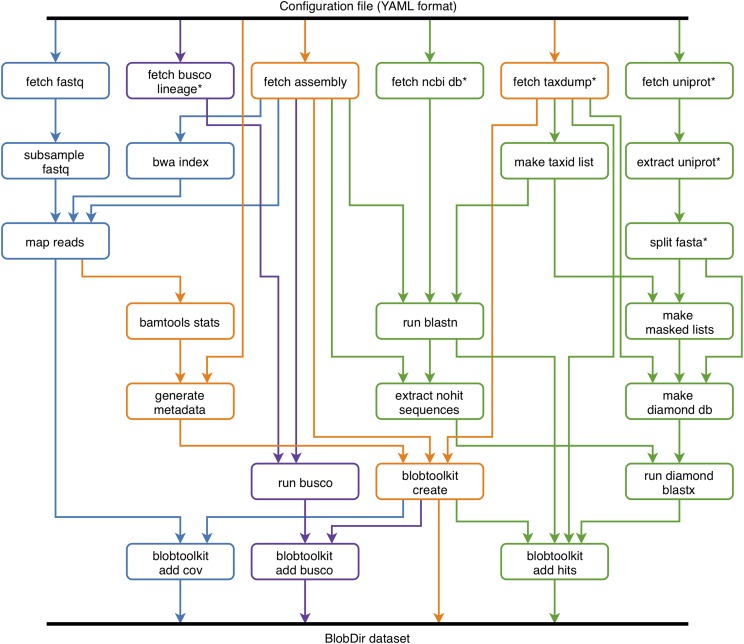
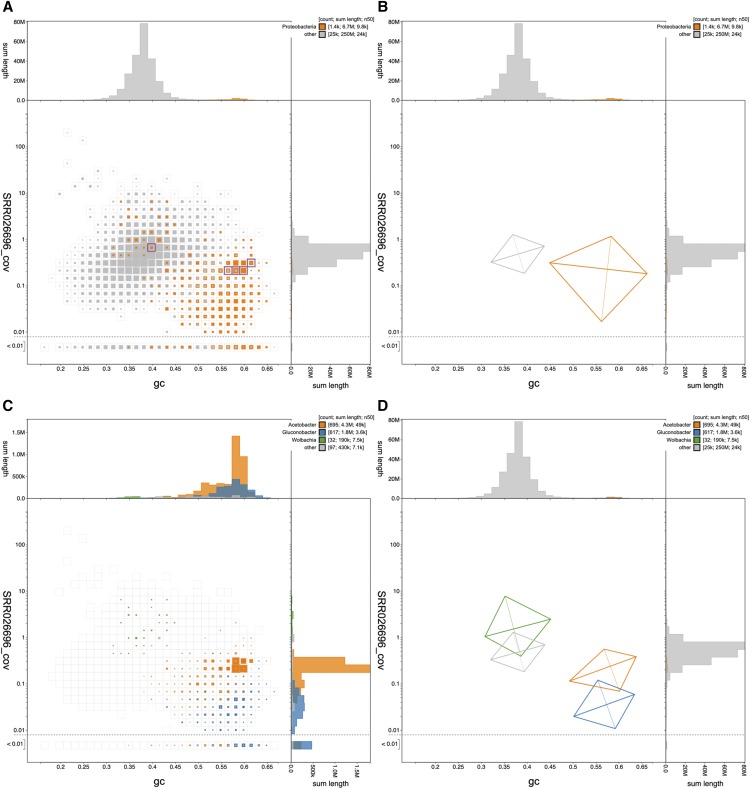
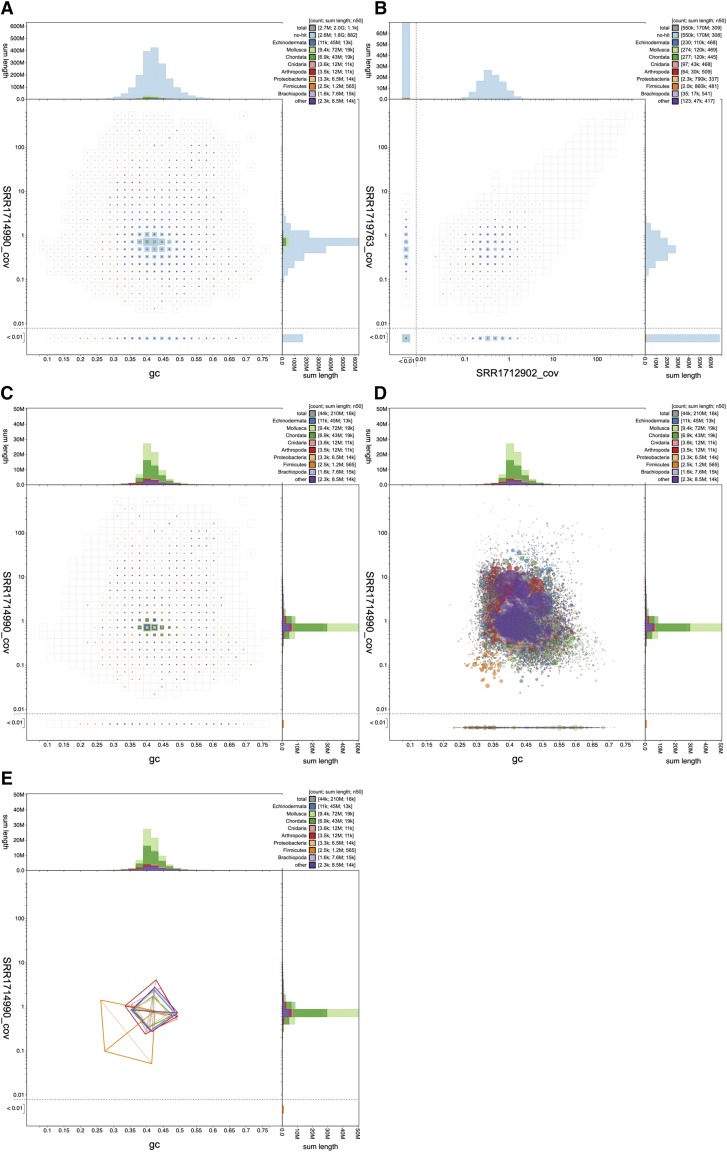
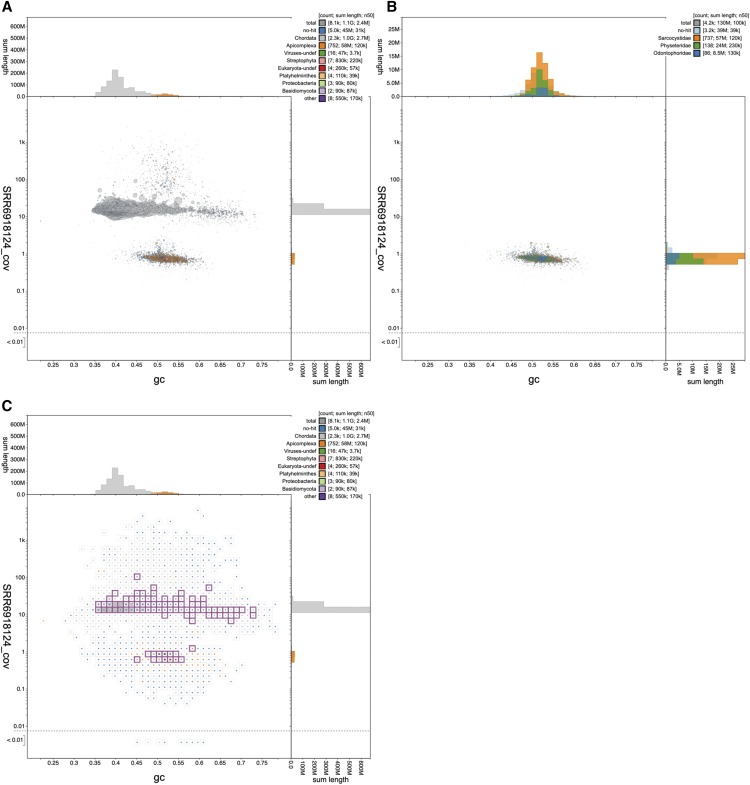
References
-
- Andreson R., Roosaare M., Kaplinski L., Laht S., Kõressaar T. et al. , 2019. Gene content of the fish-hunting cone snail Conus consors. bioRxiv. 590695 10.1101/590695 - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources