

Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke
- PMID: 32071228
- PMCID: PMC7060720
- DOI: 10.1073/pnas.1916427117
Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke
Abstract
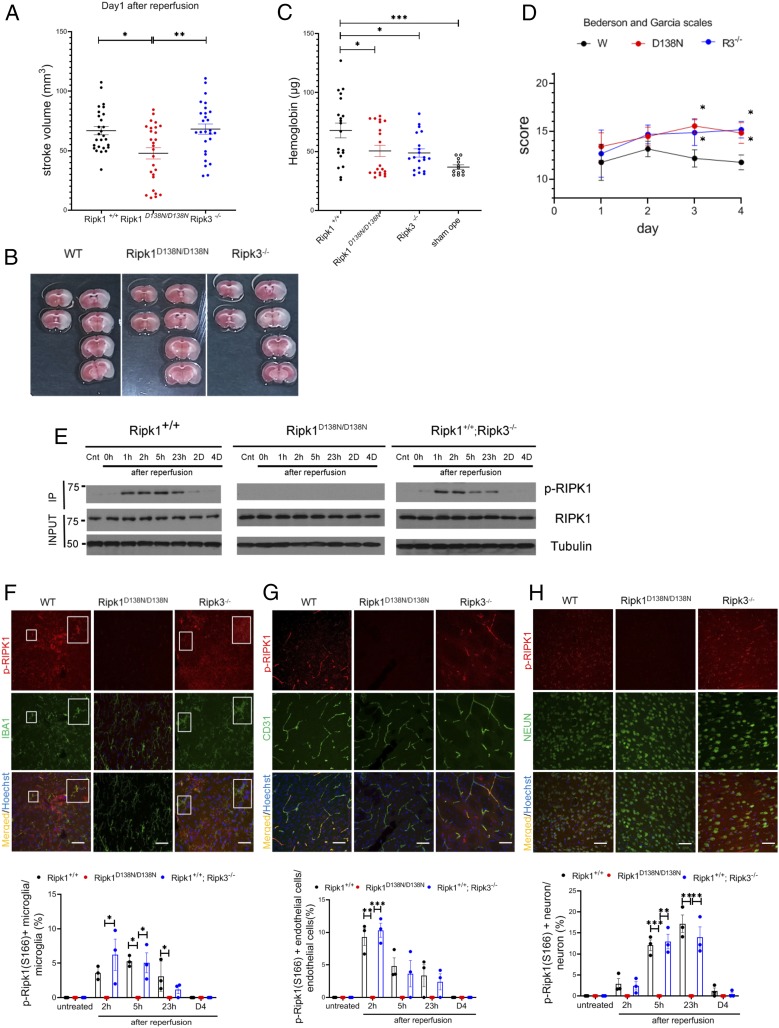
Apoptosis and necroptosis are two regulated cell death mechanisms; however, the interaction between these cell death pathways in vivo is unclear. Here we used cerebral ischemia/reperfusion as a model to investigate the interaction between apoptosis and necroptosis. We show that the activation of RIPK1 sequentially promotes necroptosis followed by apoptosis in a temporally specific manner. Cerebral ischemia/reperfusion insult rapidly activates necroptosis to promote cerebral hemorrhage and neuroinflammation. Ripk3 deficiency reduces cerebral hemorrhage and delays the onset of neural damage mediated by inflammation. Reduced cerebral perfusion resulting from arterial occlusion promotes the degradation of TAK1, a suppressor of RIPK1, and the transition from necroptosis to apoptosis. Conditional knockout of TAK1 in microglial/infiltrated macrophages and neuronal lineages sensitizes to ischemic infarction by promoting apoptosis. Taken together, our results demonstrate the critical role of necroptosis in mediating neurovascular damage and hypoperfusion-induced TAK1 loss, which subsequently promotes apoptosis and cerebral pathology in stroke and neurodegeneration.
Keywords: RIPK1; RIPK3; apoptosis; necroptosis; stroke.
Conflict of interest statement
Competing interest statement: J.Y. is a consultant of Denali Therapeutics Inc., which has licensed the necrostatin technology. M.P. received consulting and speaker fees from Genentech, GlaxoSmithKline, Boehringer Ingelheim, and Sanofi. All other authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
