

Targeting Oncogenic Super Enhancers in MYC-Dependent AML Using a Small Molecule Activator of NR4A Nuclear Receptors
- PMID: 32071334
- PMCID: PMC7029036
- DOI: 10.1038/s41598-020-59469-3
Targeting Oncogenic Super Enhancers in MYC-Dependent AML Using a Small Molecule Activator of NR4A Nuclear Receptors
Abstract
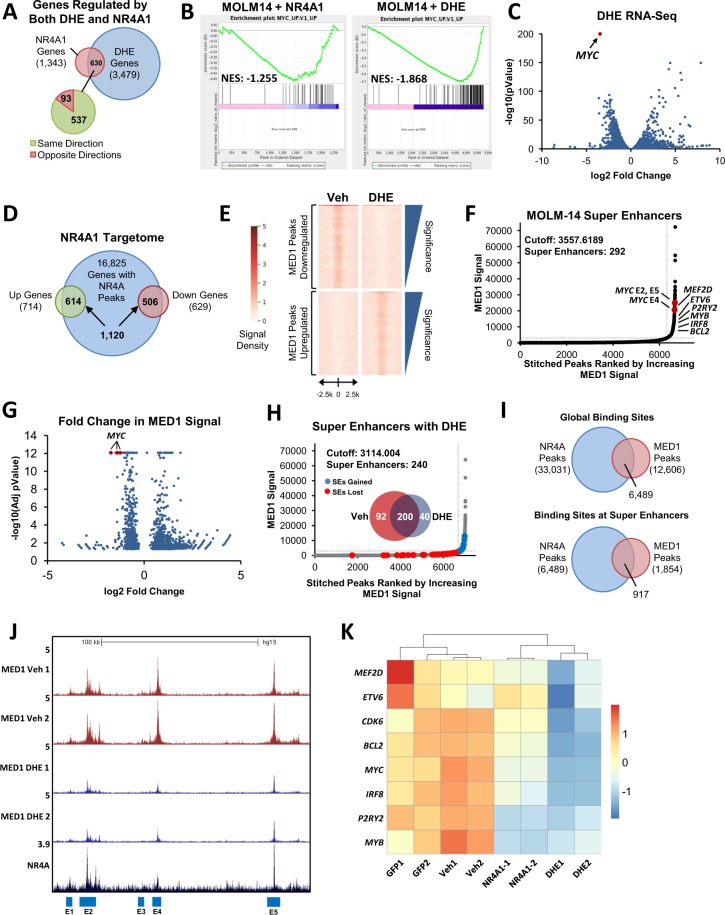
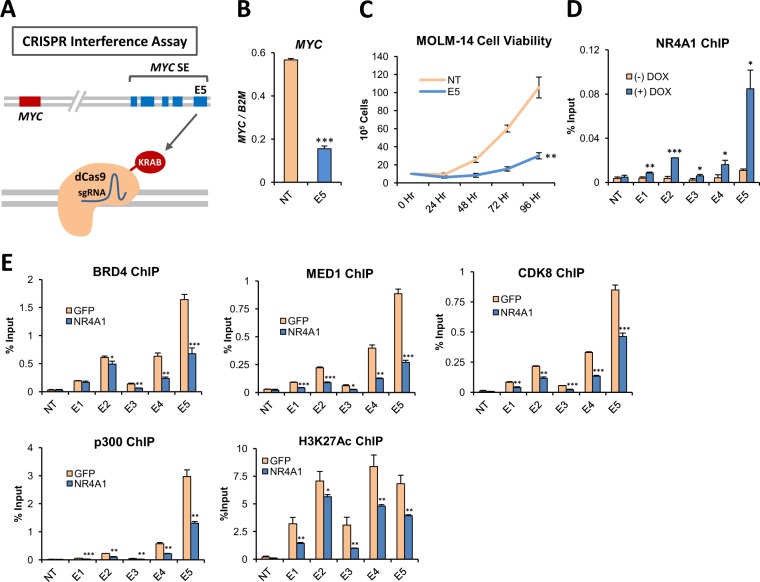
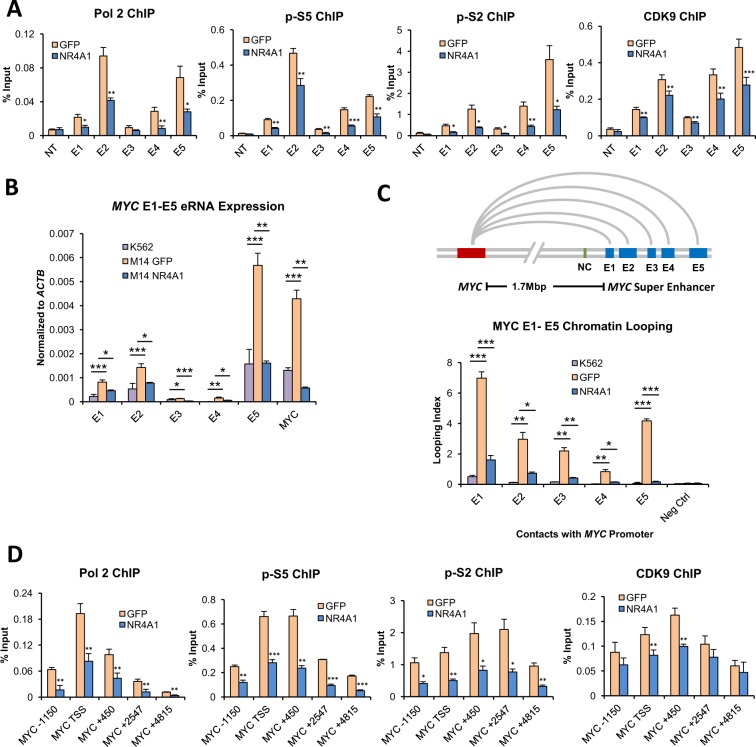
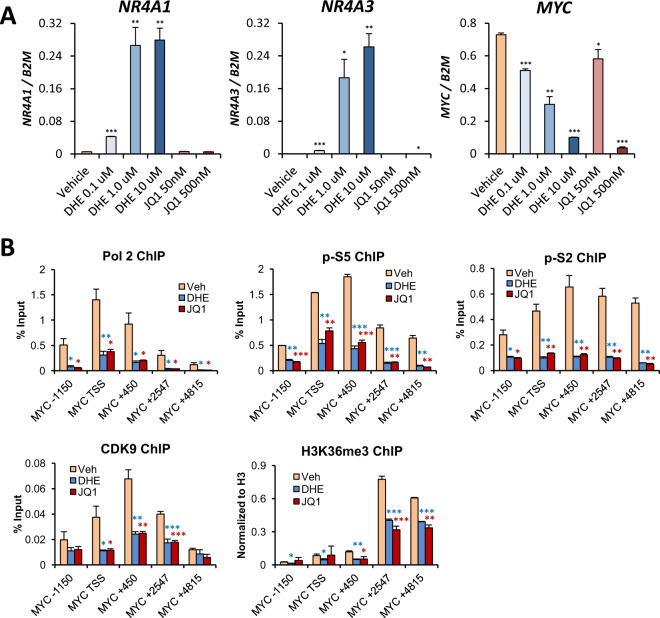
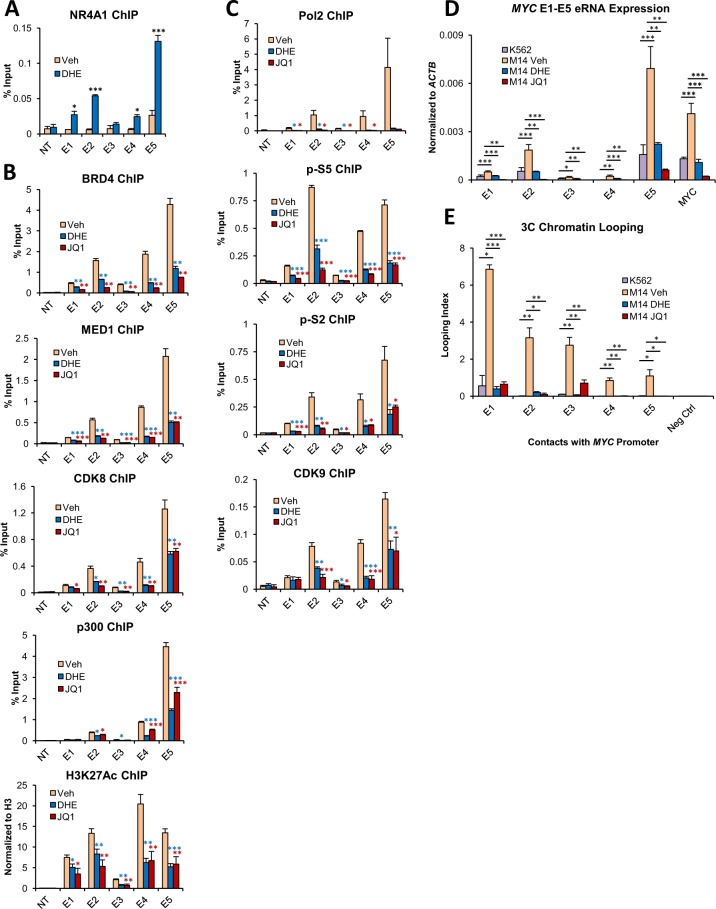
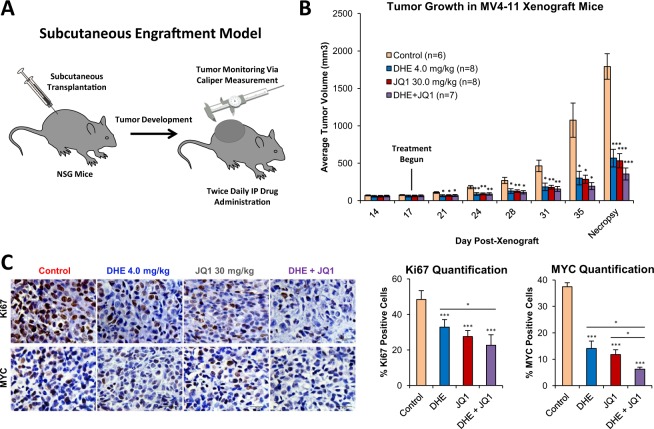
Epigenetic reprogramming in Acute Myeloid Leukemia (AML) leads to the aberrant activation of super enhancer (SE) landscapes that drive the expression of key oncogenes, including the oncogenic MYC pathway. These SEs have been identified as promising therapeutic targets, and have given rise to a new class of drugs, including BET protein inhibitors, which center on targeting SE activity. NR4A nuclear receptors are tumor suppressors of AML that function in part through transcriptional repression of the MYC-driven oncogenic program via mechanisms that remain unclear. Here we show that NR4A1, and the NR4A inducing drug dihydroergotamine (DHE), regulate overlapping gene expression programs in AML and repress transcription of a subset of SE-associated leukemic oncogenes, including MYC. NR4As interact with an AML-selective SE cluster that governs MYC transcription and decommissions its activation status by dismissing essential SE-bound coactivators including BRD4, Mediator and p300, leading to loss of p300-dependent H3K27 acetylation and Pol 2-dependent eRNA transcription. DHE shows similar efficacy to the BET inhibitor JQ1 at repressing SE-dependent MYC expression and AML growth in mouse xenografts. Thus, DHE induction of NR4As provides an alternative strategy to BET inhibitors to target MYC dependencies via suppression of the AML-selective SE governing MYC expression.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
