

Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death
- PMID: 32071389
- PMCID: PMC7028984
- DOI: 10.1038/s42003-020-0792-9
Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death
Abstract
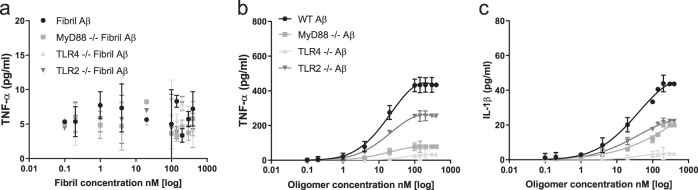
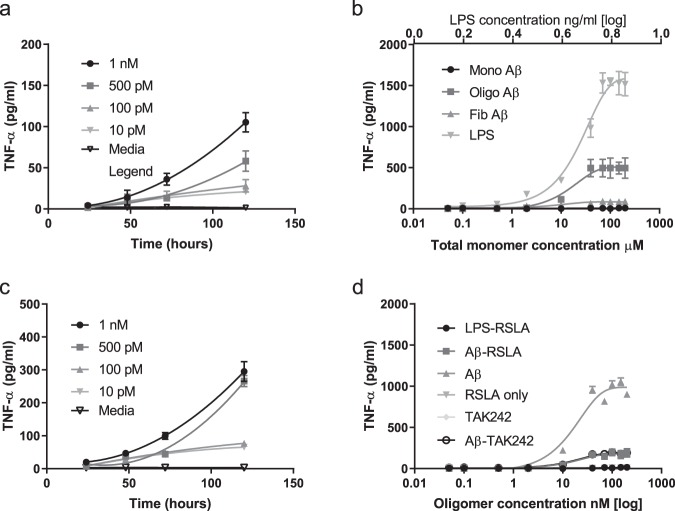
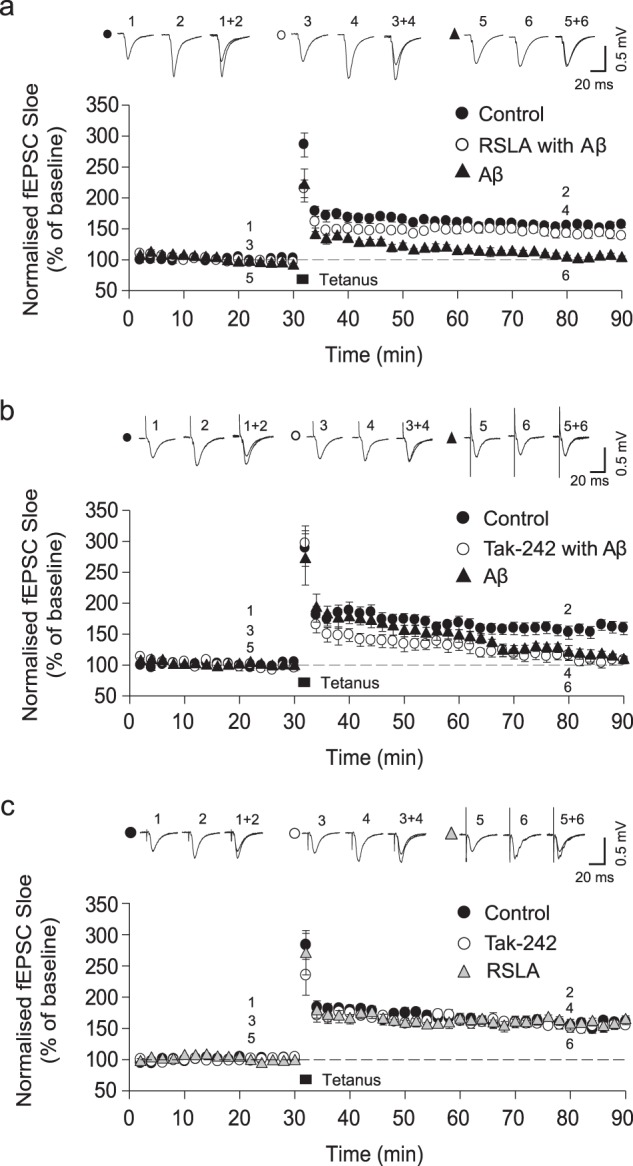
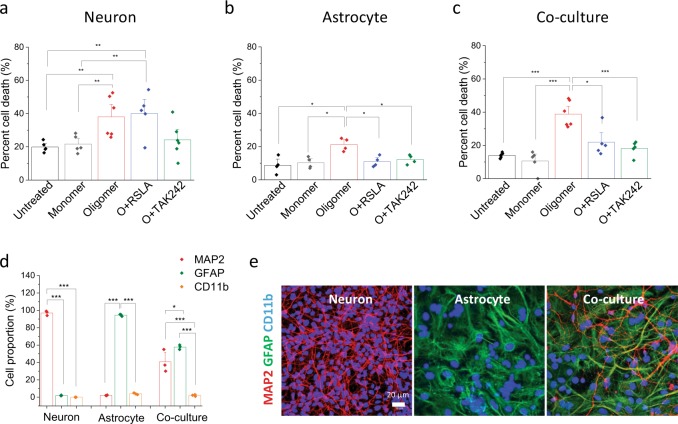
The molecular events causing memory loss and neuronal cell death in Alzheimer's disease (AD) over time are still unknown. Here we found that picomolar concentrations of soluble oligomers of synthetic beta amyloid (Aβ42) aggregates incubated with BV2 cells or rat astrocytes caused a sensitised response of Toll-like receptor 4 (TLR4) with time, leading to increased production of TNF-α. Aβ aggregates caused long term potentiation (LTP) deficit in hippocampal slices and predominantly neuronal cell death in co-cultures of astrocytes and neurons, which was blocked by TLR4 antagonists. Soluble Aβ aggregates cause LTP deficit and neuronal death via an autocrine/paracrine mechanism due to TLR4 signalling. These findings suggest that the TLR4-mediated inflammatory response may be a key pathophysiological process in AD.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
