

Inhibition of Bromodomain and Extraterminal Domain (BET) Proteins by JQ1 Unravels a Novel Epigenetic Modulation to Control Lipid Homeostasis
- PMID: 32075110
- PMCID: PMC7072965
- DOI: 10.3390/ijms21041297
Inhibition of Bromodomain and Extraterminal Domain (BET) Proteins by JQ1 Unravels a Novel Epigenetic Modulation to Control Lipid Homeostasis
Abstract
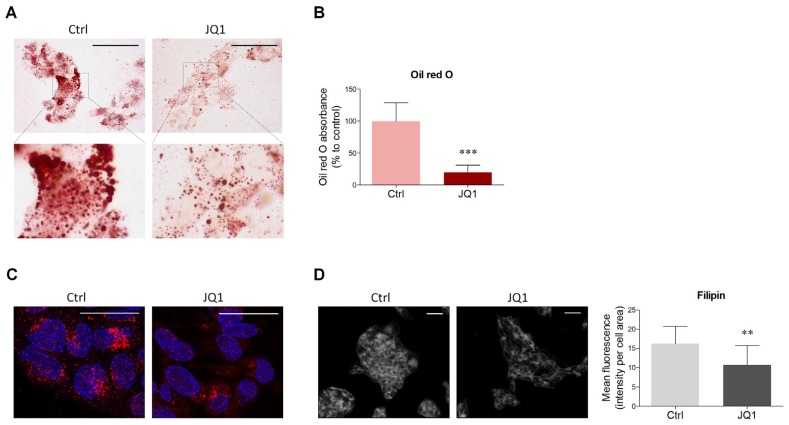
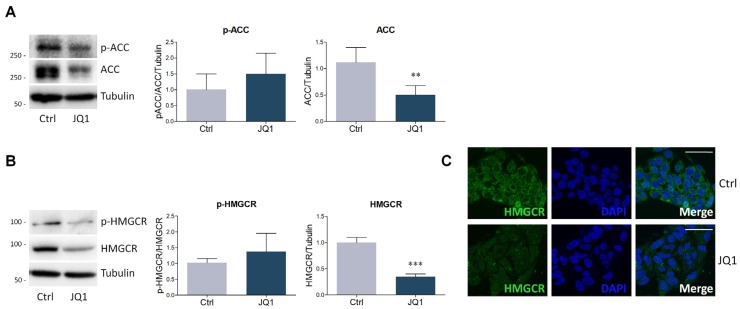
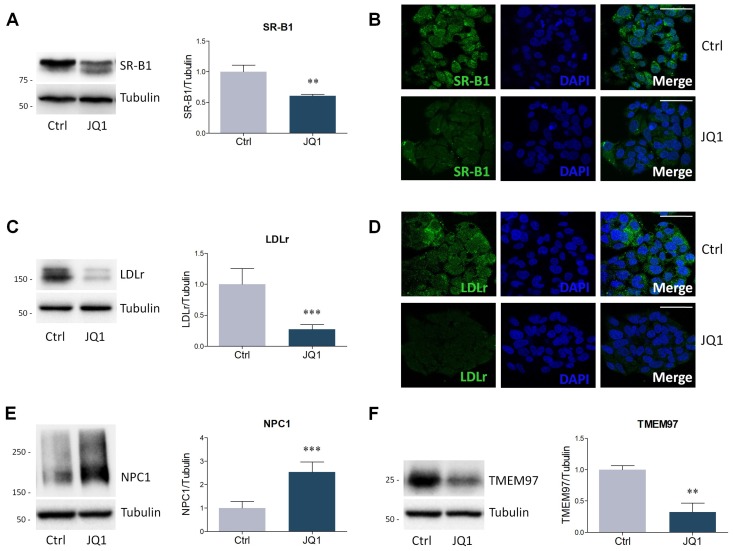
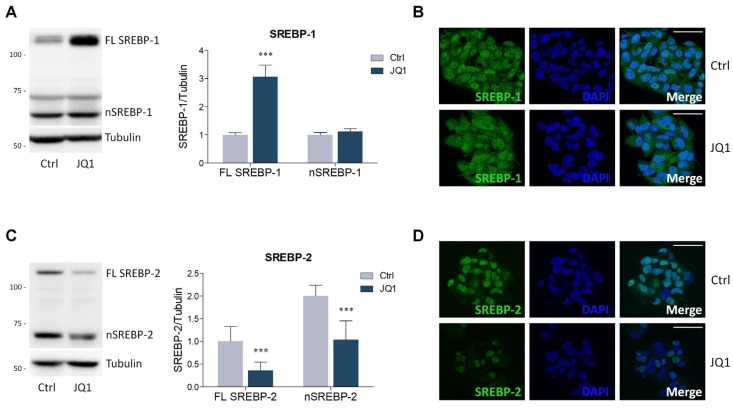
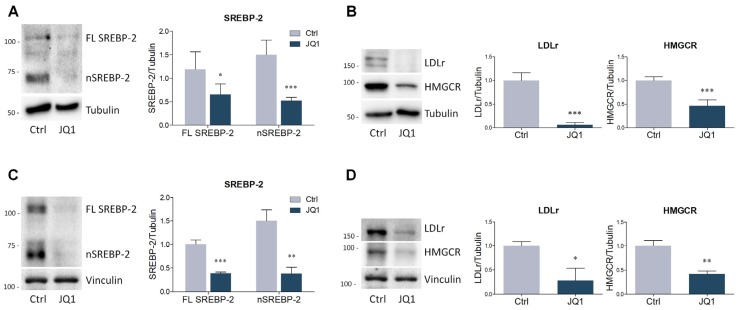
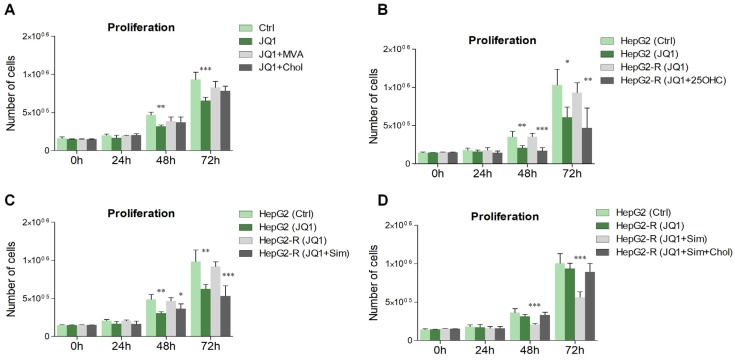
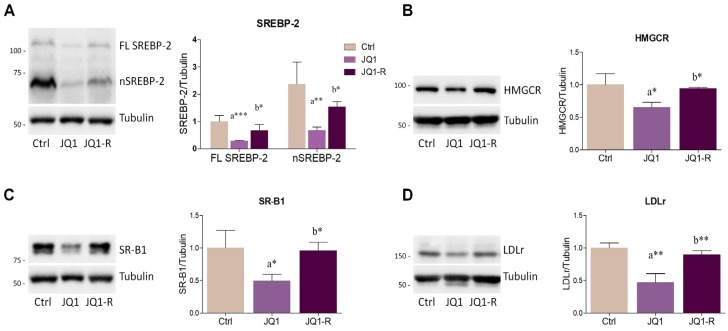
The homeostatic control of lipid metabolism is essential for many fundamental physiological processes. A deep understanding of its regulatory mechanisms is pivotal to unravel prospective physiopathological factors and to identify novel molecular targets that could be employed to design promising therapies in the management of lipid disorders. Here, we investigated the role of bromodomain and extraterminal domain (BET) proteins in the regulation of lipid metabolism. To reach this aim, we used a loss-of-function approach by treating HepG2 cells with JQ1, a powerful and selective BET inhibitor. The main results demonstrated that BET inhibition by JQ1 efficiently decreases intracellular lipid content, determining a significant modulation of proteins involved in lipid biosynthesis, uptake and intracellular trafficking. Importantly, the capability of BET inhibition to slow down cell proliferation is dependent on the modulation of cholesterol metabolism. Taken together, these data highlight a novel epigenetic mechanism involved in the regulation of lipid homeostasis.
Keywords: BET proteins; HMGCR; JQ1; LDLr; SREBP; TMEM97; cell proliferation; cholesterol; epigenetics; lipid metabolism.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Chen H.W. Role of cholesterol metabolism in cell growth. Fed. Proc. 1984;43:126–130. - PubMed
-
- Yao C.H., Fowle-Grider R., Mahieu N.G., Liu G.Y., Chen Y.J., Wang R., Singh M., Potter G.S., Gross R.W., Schaefer J., et al. Exogenous Fatty Acids Are the Preferred Source of Membrane Lipids in Proliferating Fibroblasts. Cell Chem. Biol. 2016;23:483–493. doi: 10.1016/j.chembiol.2016.03.007. - DOI - PMC - PubMed
