

Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies
- PMID: 32075145
- PMCID: PMC7074327
- DOI: 10.3390/jcm9020520
Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies
Abstract
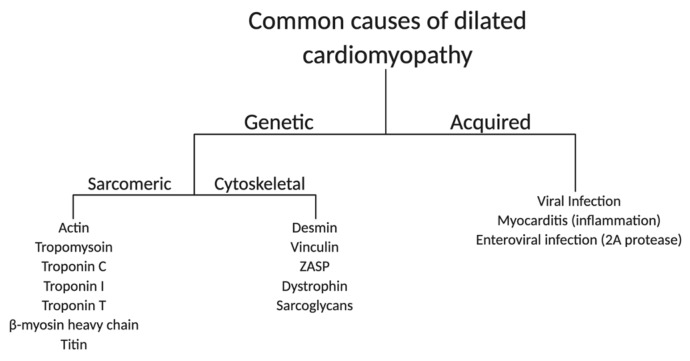
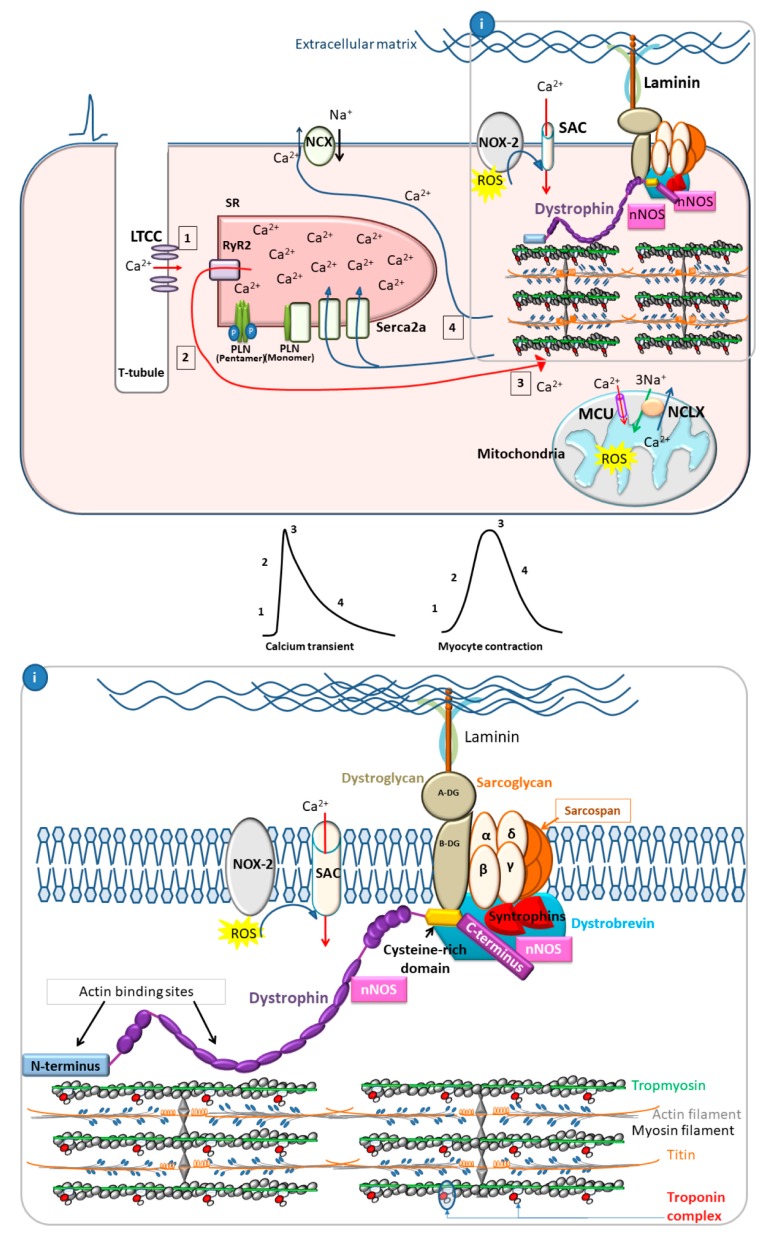
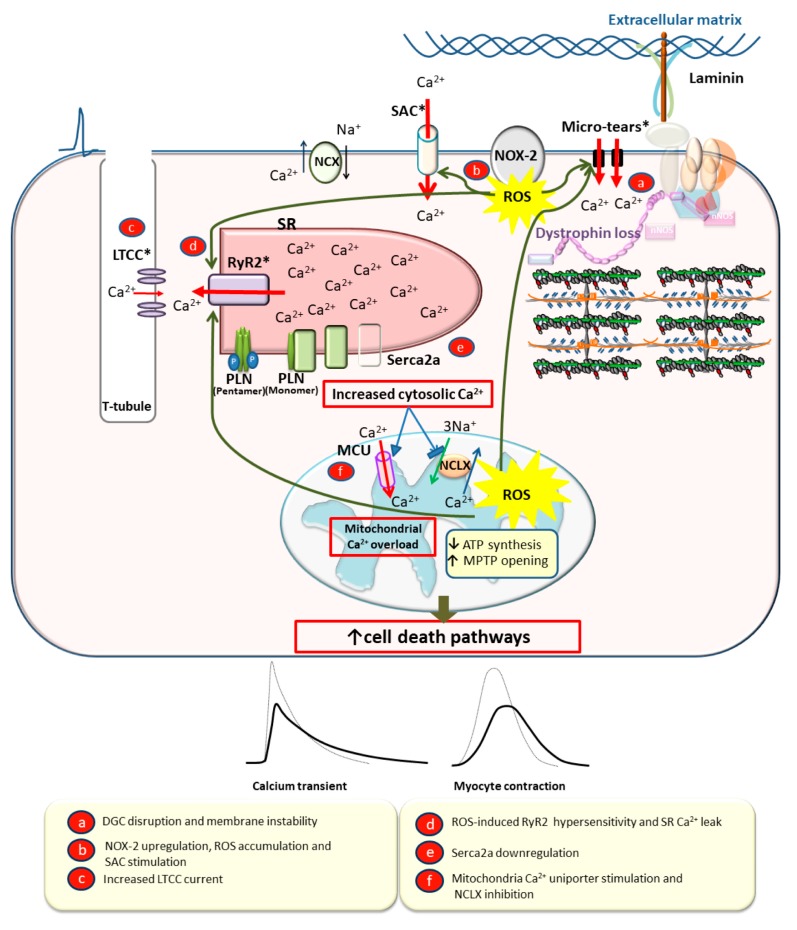
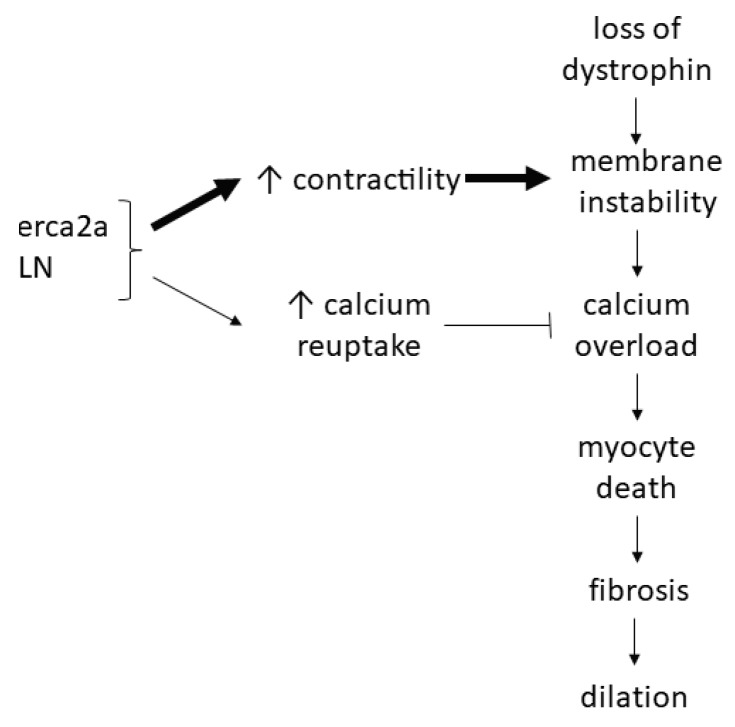
: Duchenne muscular dystrophy (DMD) is an X-linked recessive disease resulting in the loss of dystrophin, a key cytoskeletal protein in the dystrophin-glycoprotein complex. Dystrophin connects the extracellular matrix with the cytoskeleton and stabilizes the sarcolemma. Cardiomyopathy is prominent in adolescents and young adults with DMD, manifesting as dilated cardiomyopathy (DCM) in the later stages of disease. Sarcolemmal instability, leading to calcium mishandling and overload in the cardiac myocyte, is a key mechanistic contributor to muscle cell death, fibrosis, and diminished cardiac contractile function in DMD patients. Current therapies for DMD cardiomyopathy can slow disease progression, but they do not directly target aberrant calcium handling and calcium overload. Experimental therapeutic targets that address calcium mishandling and overload include membrane stabilization, inhibition of stretch-activated channels, ryanodine receptor stabilization, and augmentation of calcium cycling via modulation of the Serca2a/phospholamban (PLN) complex or cytosolic calcium buffering. This paper addresses what is known about the mechanistic basis of calcium mishandling in DCM, with a focus on DMD cardiomyopathy. Additionally, we discuss currently utilized therapies for DMD cardiomyopathy, and review experimental therapeutic strategies targeting the calcium handling defects in DCM and DMD cardiomyopathy.
Keywords: Serca2a; calcium; dilated cardiomyopathy; gene therapy; heart; mdx; membrane stabilization; muscular dystrophy; oxidative stress; phospholamban.
Conflict of interest statement
The authors do not declare any conflict of interest.
Figures
Similar articles
-
Exacerbation of dystrophic cardiomyopathy by phospholamban deficiency mediated chronically increased cardiac Ca2+ cycling in vivo.Am J Physiol Heart Circ Physiol. 2018 Dec 1;315(6):H1544-H1552. doi: 10.1152/ajpheart.00341.2018. Epub 2018 Aug 17. Am J Physiol Heart Circ Physiol. 2018. PMID: 30118340 Free PMC article.
-
Ryanodine receptor dysfunction causes senescence and fibrosis in Duchenne dilated cardiomyopathy.J Cachexia Sarcopenia Muscle. 2024 Apr;15(2):536-551. doi: 10.1002/jcsm.13411. Epub 2024 Jan 14. J Cachexia Sarcopenia Muscle. 2024. PMID: 38221511 Free PMC article.
-
Intracellular calcium handling in ventricular myocytes from mdx mice.Am J Physiol Heart Circ Physiol. 2007 Feb;292(2):H846-55. doi: 10.1152/ajpheart.00688.2006. Epub 2006 Sep 29. Am J Physiol Heart Circ Physiol. 2007. PMID: 17012353
-
The multifaceted view of heart problem in Duchenne muscular dystrophy.Cell Mol Life Sci. 2021 Jul;78(14):5447-5468. doi: 10.1007/s00018-021-03862-2. Epub 2021 Jun 6. Cell Mol Life Sci. 2021. PMID: 34091693 Free PMC article. Review.
-
Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies.Front Physiol. 2021 Apr 9;12:647010. doi: 10.3389/fphys.2021.647010. eCollection 2021. Front Physiol. 2021. PMID: 33897454 Free PMC article. Review.
Cited by
-
Anti-Fibrotic Effect of Human Wharton's Jelly-Derived Mesenchymal Stem Cells on Skeletal Muscle Cells, Mediated by Secretion of MMP-1.Int J Mol Sci. 2020 Aug 29;21(17):6269. doi: 10.3390/ijms21176269. Int J Mol Sci. 2020. PMID: 32872523 Free PMC article.
-
Non-Cardiac Cause of Death in Selected Group Children with Cardiac Pathology: A Retrospective Single Institute Study.Children (Basel). 2022 Mar 2;9(3):335. doi: 10.3390/children9030335. Children (Basel). 2022. PMID: 35327707 Free PMC article.
-
Cardiac Remodeling in Cancer-Induced Cachexia: Functional, Structural, and Metabolic Contributors.Cells. 2022 Jun 15;11(12):1931. doi: 10.3390/cells11121931. Cells. 2022. PMID: 35741060 Free PMC article. Review.
-
Anti-Remodeling Cardiac Therapy in Patients With Duchenne Muscular Dystrophy, Meta-Analysis Study.Front Pharmacol. 2022 Jan 20;12:769896. doi: 10.3389/fphar.2021.769896. eCollection 2021. Front Pharmacol. 2022. PMID: 35126112 Free PMC article. Review.
-
A Hypothesized Therapeutic Role of (Z)-Endoxifen in Duchenne Muscular Dystrophy (DMD).Degener Neurol Neuromuscul Dis. 2025 Mar 15;15:1-15. doi: 10.2147/DNND.S496904. eCollection 2025. Degener Neurol Neuromuscul Dis. 2025. PMID: 40124418 Free PMC article.
References
-
- World Health Organization . World Heath Statistics 2017: Monitoring Health for the SDGs, Sustainable Development Goals. World Health Organization; Geneva, Switzerland: 2017.
-
- Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R., et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
