

Stim1 Polymorphism Disrupts Immune Signaling and Creates Renal Injury in Hypertension
- PMID: 32075490
- PMCID: PMC7335582
- DOI: 10.1161/JAHA.119.014142
Stim1 Polymorphism Disrupts Immune Signaling and Creates Renal Injury in Hypertension
Abstract
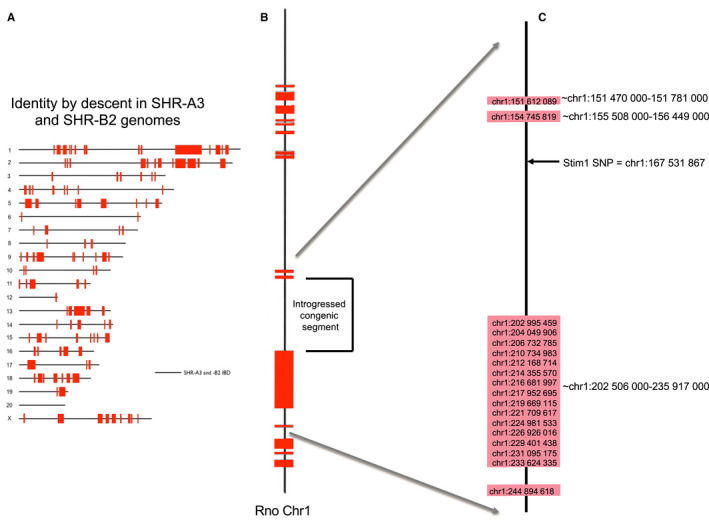
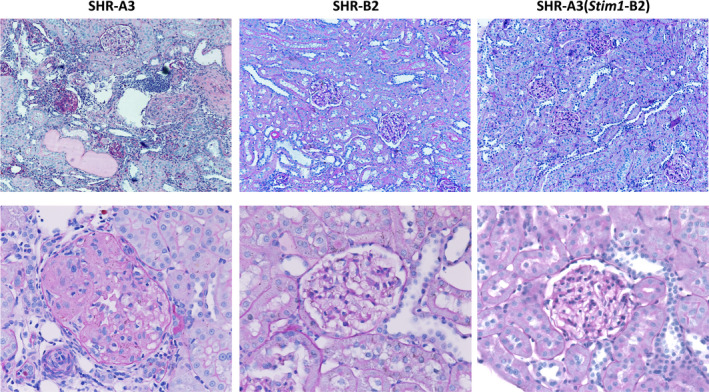
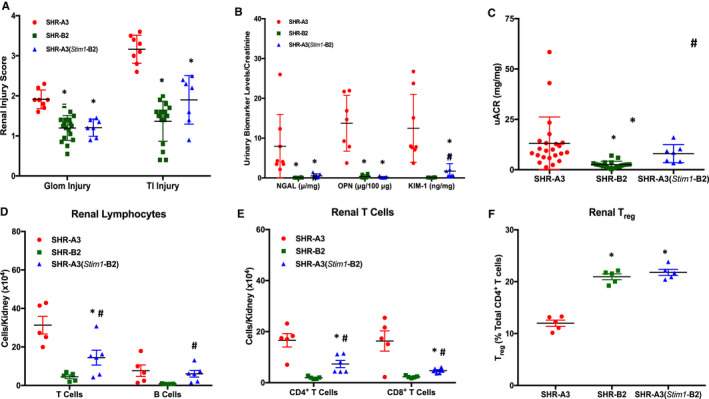
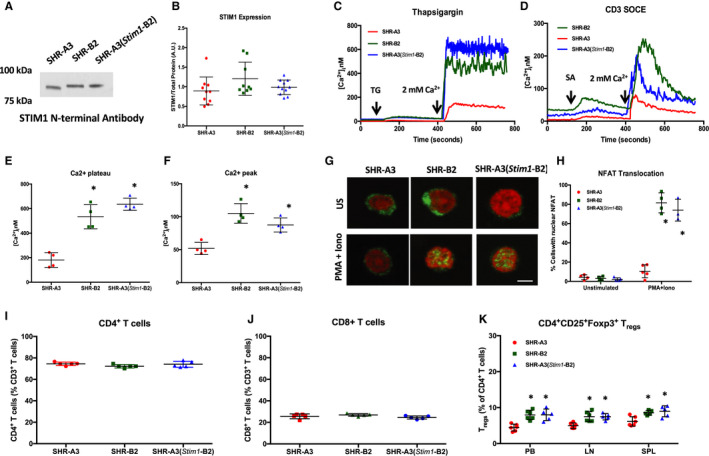
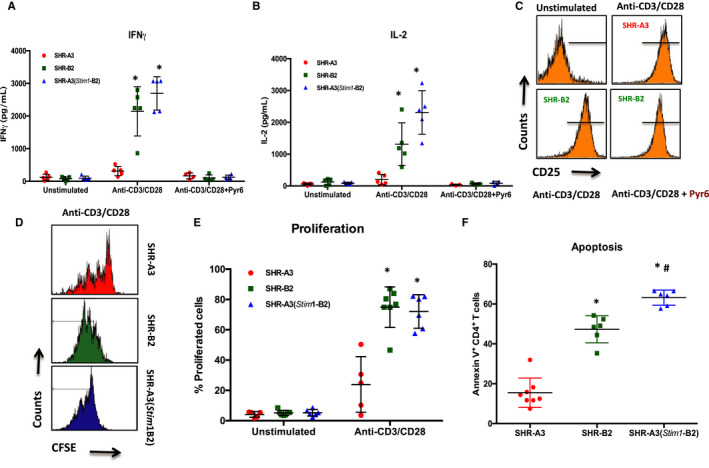
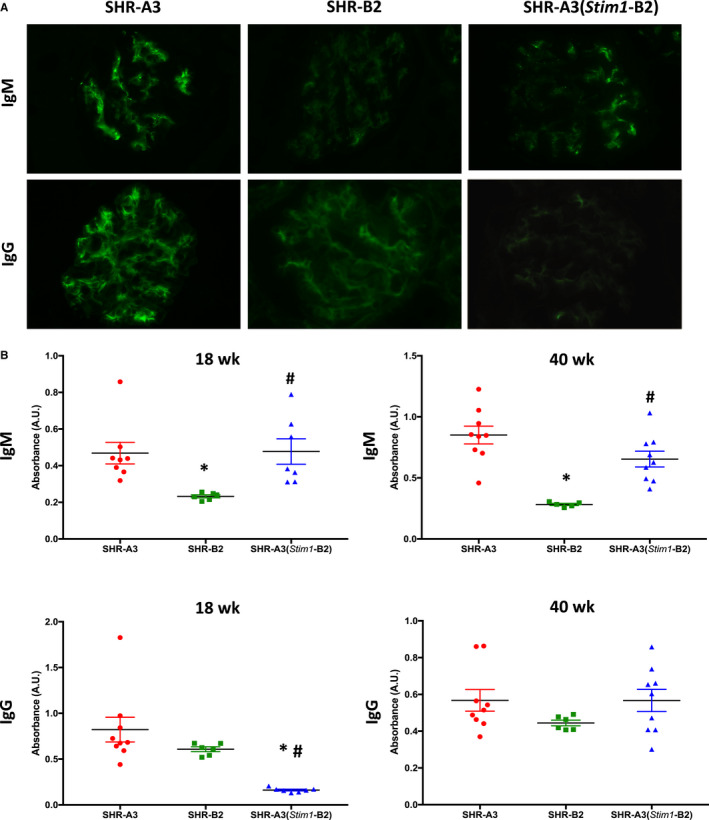
Background Spontaneously hypertensive rats of the stroke-prone line (SHR-A3) develop hypertensive renal disease as a result of naturally occurring genetic variation. Our prior work identified a single-nucleotide polymorphism unique to SHR-A3 that results in truncation of the carboxy terminus of STIM1. The SHR-B2 line, which is also hypertensive but resists hypertensive renal injury, expresses the wild-type STIM1. STIM1 plays a central role in lymphocyte calcium signaling that directs immune effector responses. Here we show that major defects in lymphocyte function affecting calcium signaling, nuclear factor of activated T cells activation, cytokine production, proliferation, apoptosis, and regulatory T-cell development are present in SHR-A3 and attributable to STIM1. Methods and Results To assess the role of Stim1 variation in susceptibility to hypertensive renal injury, we created a Stim1 congenic line, SHR-A3(Stim1-B2), and STIM1 function was rescued in SHR-A3. We found that Stim1 gene rescue restores disturbed lymphocyte function in SHR-A3. Hypertensive renal injury was compared in SHR-A3 and the SHR-A3(Stim1-B2) congenic line. Histologically assessed renal injury was markedly reduced in SHR-A3(Stim1-B2), as were renal injury biomarker levels measured in urine. Stim1 deficiency has been linked to the emergence of antibody-mediated autoimmunity. Renal glomerular immunoglobulin deposition was greater in SHR-A3 than SHR-B2 and was reduced by Stim1 congenic substitution. Serum anti-double-stranded DNA antibody titers in SHR-A3 were elevated compared with SHR-B2 and were reduced in SHR-A3(Stim1-B2). Conclusions Stim1 deficiency in lymphocyte function originating from Stim1 truncation in SHR-A3 combines with hypertension to create end organ disease and may do so as a result of antibody formation.
Keywords: autoimmunity; hypertension; immunoglobulin; renal disease; spontaneously hypertensive rat.
Figures
References
-
- Freedman BI, Soucie JM, McClellan WM. Family history of end‐stage renal disease among incident dialysis patients. J Am Soc Nephrol. 1997;8:1942–1945. - PubMed
-
- Satko SG, Sedor JR, Iyengar SK, Freedman BI. Familial clustering of chronic kidney disease. Semin Dial. 2007;20:229–236. - PubMed
-
- Freedman BI, Spray BJ, Tuttle AB, Buckalew VM Jr. The familial risk of end‐stage renal disease in African Americans. Am J Kidney Dis. 1993;21:387–393. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
