

The African Swine Fever Virus Transcriptome
- PMID: 32075923
- PMCID: PMC7163114
- DOI: 10.1128/JVI.00119-20
The African Swine Fever Virus Transcriptome
Abstract
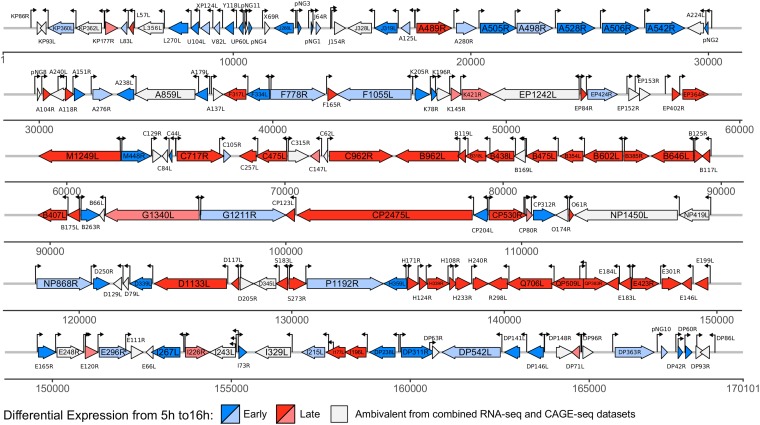
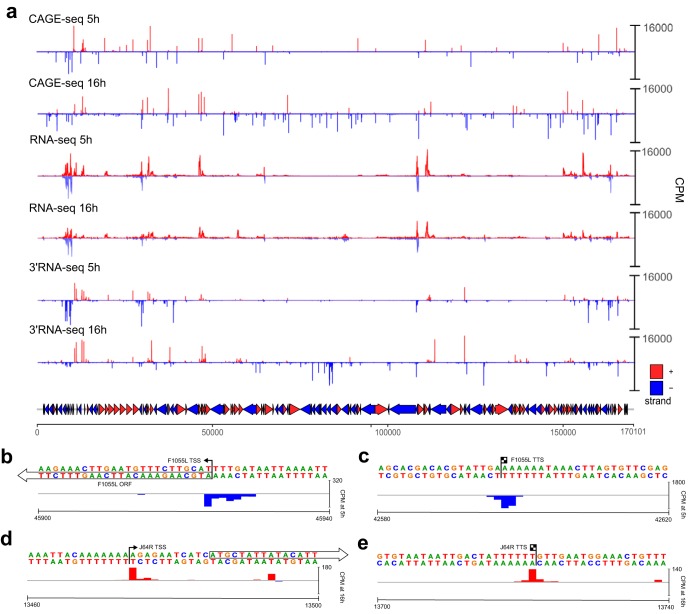
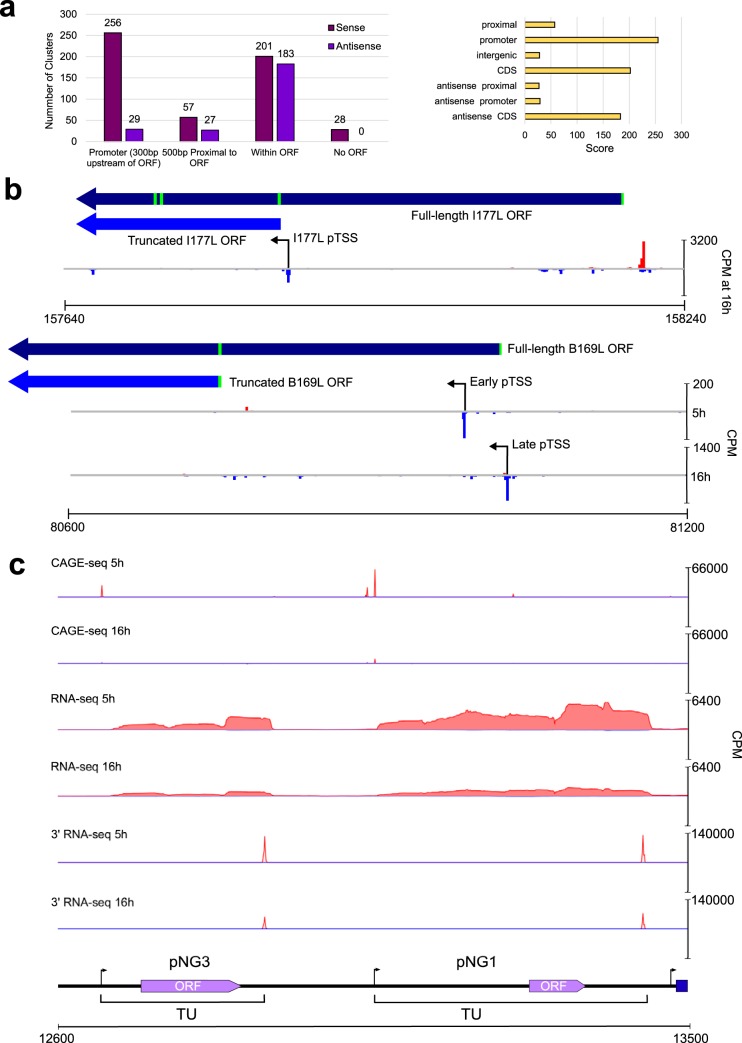
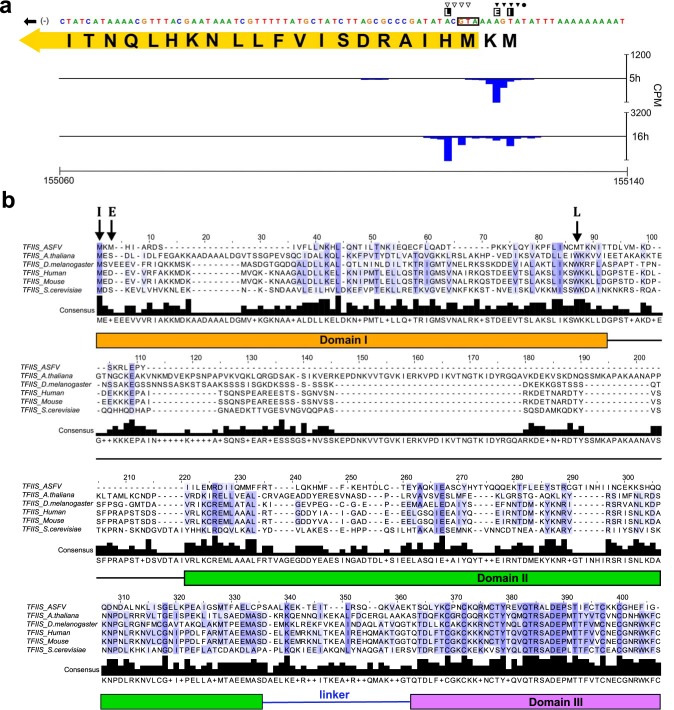
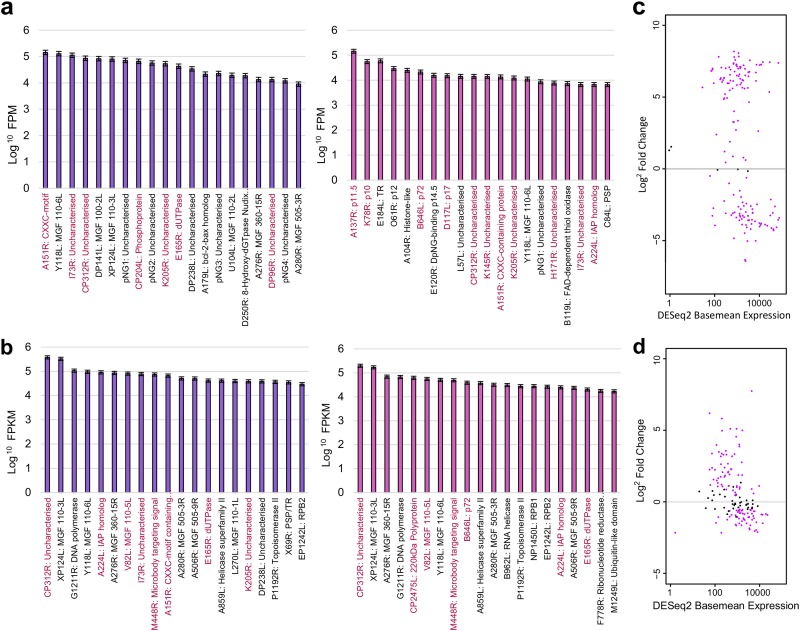
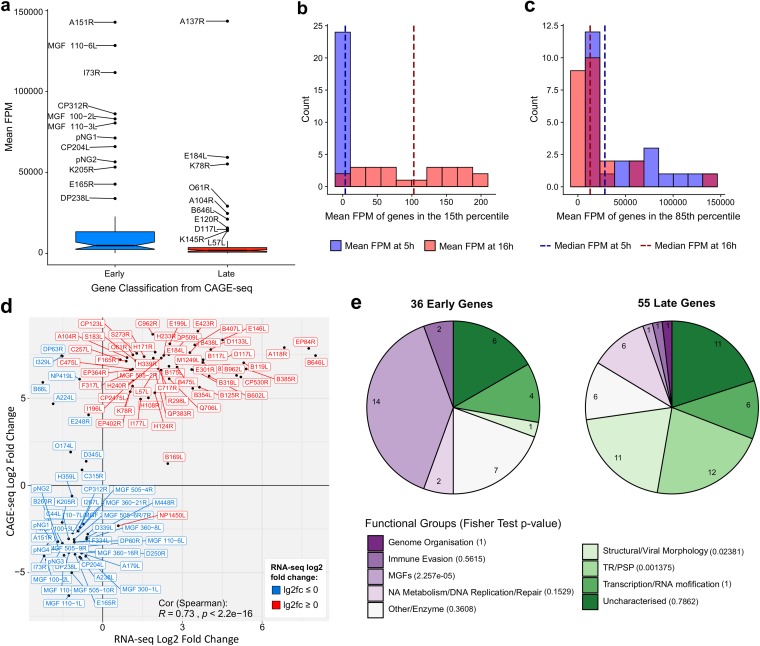
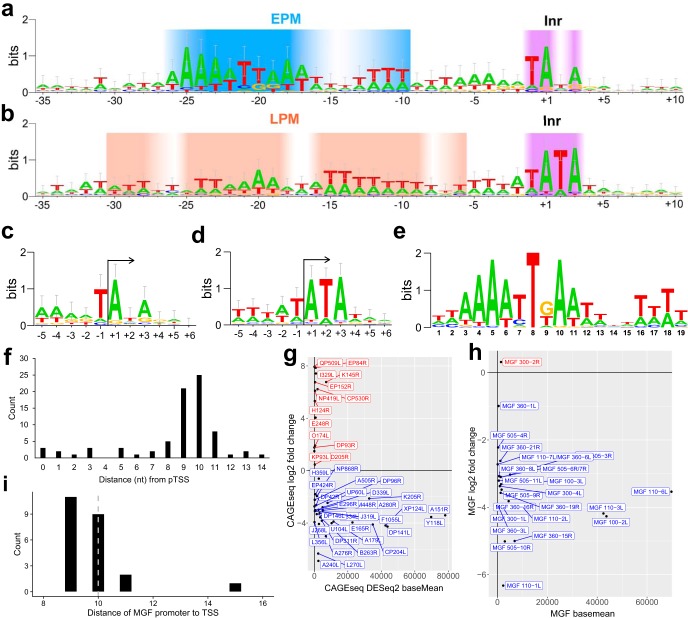
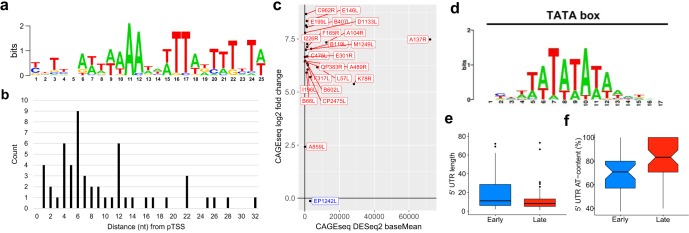
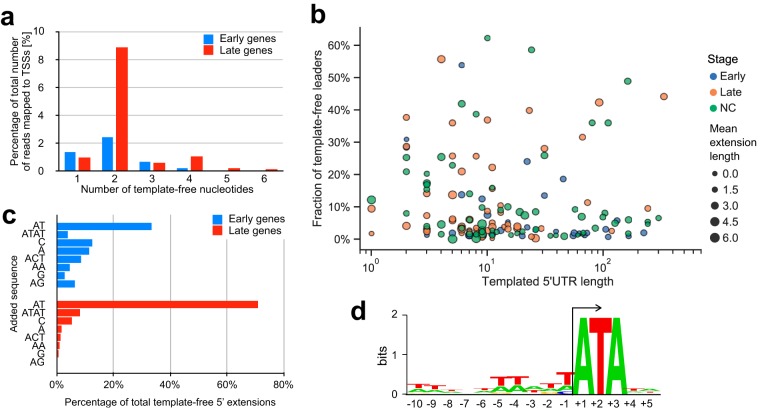
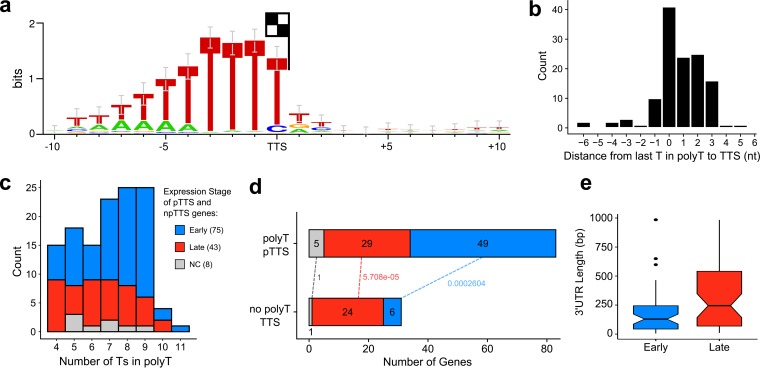
African swine fever virus (ASFV) causes hemorrhagic fever in domestic pigs, presenting the biggest global threat to animal farming in recorded history. Despite the importance of ASFV, little is known about the mechanisms and regulation of ASFV transcription. Using RNA sequencing methods, we have determined total RNA abundance, transcription start sites, and transcription termination sites at single-nucleotide resolution. This allowed us to characterize DNA consensus motifs of early and late ASFV core promoters, as well as a polythymidylate sequence determinant for transcription termination. Our results demonstrate that ASFV utilizes alternative transcription start sites between early and late stages of infection and that ASFV RNA polymerase (RNAP) undergoes promoter-proximal transcript slippage at 5' ends of transcription units, adding quasitemplated AU- and AUAU-5' extensions to mRNAs. Here, we present the first much-needed genome-wide transcriptome study that provides unique insight into ASFV transcription and serves as a resource to aid future functional analyses of ASFV genes which are essential to combat this devastating disease.IMPORTANCE African swine fever virus (ASFV) causes incurable and often lethal hemorrhagic fever in domestic pigs. In 2020, ASF presents an acute and global animal health emergency that has the potential to devastate entire national economies as effective vaccines or antiviral drugs are not currently available (according to the Food and Agriculture Organization of the United Nations). With major outbreaks ongoing in Eastern Europe and Asia, urgent action is needed to advance our knowledge about the fundamental biology of ASFV, including the mechanisms and temporal control of gene expression. A thorough understanding of RNAP and transcription factor function, and of the sequence context of their promoter motifs, as well as accurate knowledge of which genes are expressed when and the amino acid sequence of the encoded proteins, is direly needed for the development of antiviral drugs and vaccines.
Keywords: African swine fever virus; NCLDV; RNA polymerases; RNA-seq; gene expression; promoters; transcription; transcription start site; virology; zoonotic infections.
Copyright © 2020 Cackett et al.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- WT 108877/B/15/Z/WT_/Wellcome Trust/United Kingdom
- BBS/E/I/00007034/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- WT 207446/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- BBS/E/I/0007030/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
