

Cellular deconvolution of GTEx tissues powers discovery of disease and cell-type associated regulatory variants
- PMID: 32075962
- PMCID: PMC7031340
- DOI: 10.1038/s41467-020-14561-0
Cellular deconvolution of GTEx tissues powers discovery of disease and cell-type associated regulatory variants
Erratum in
-
Author Correction: Cellular deconvolution of GTEx tissues powers discovery of disease and cell-type associated regulatory variants.Nat Commun. 2020 Sep 1;11(1):4426. doi: 10.1038/s41467-020-18341-8. Nat Commun. 2020. PMID: 32873812 Free PMC article.
Abstract
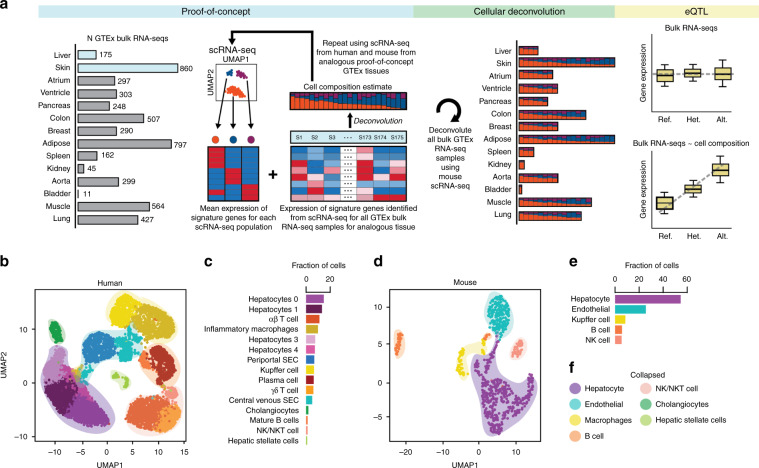
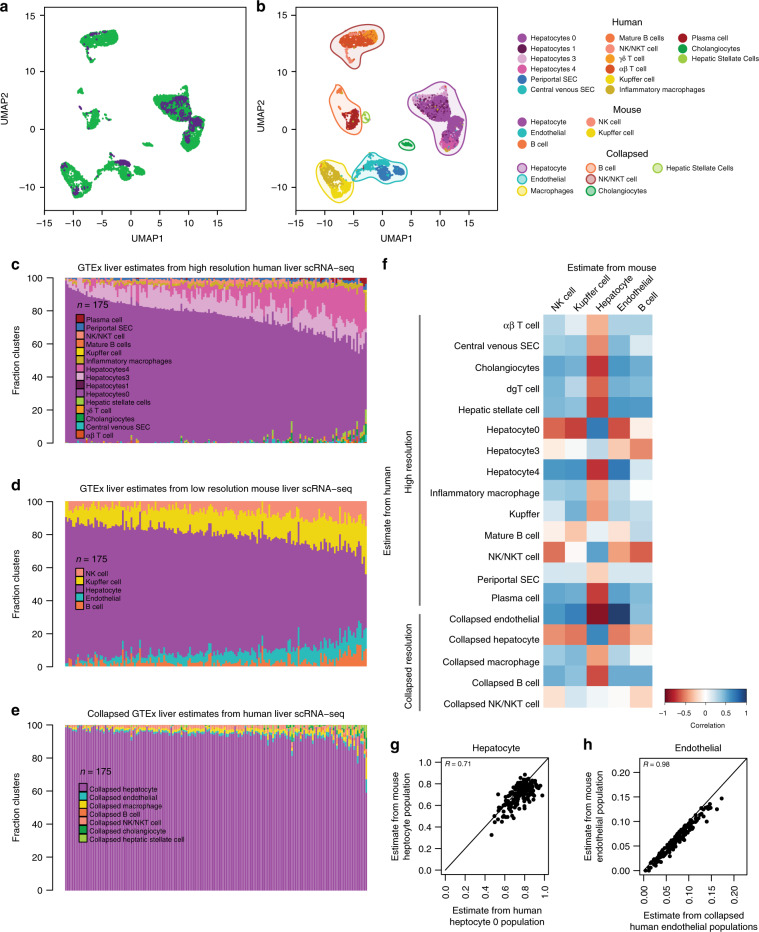
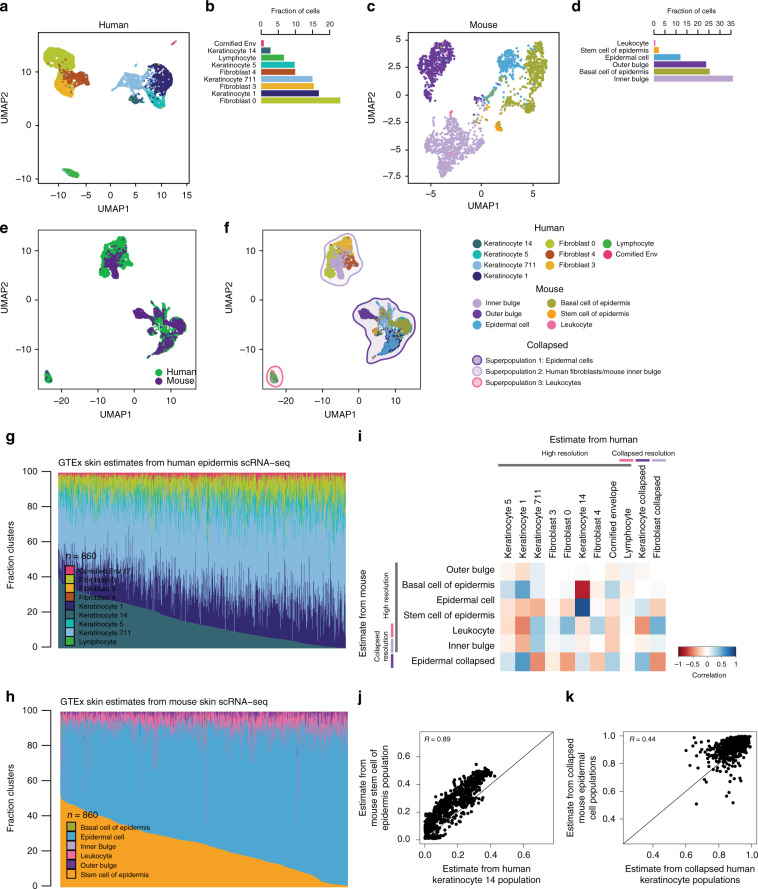
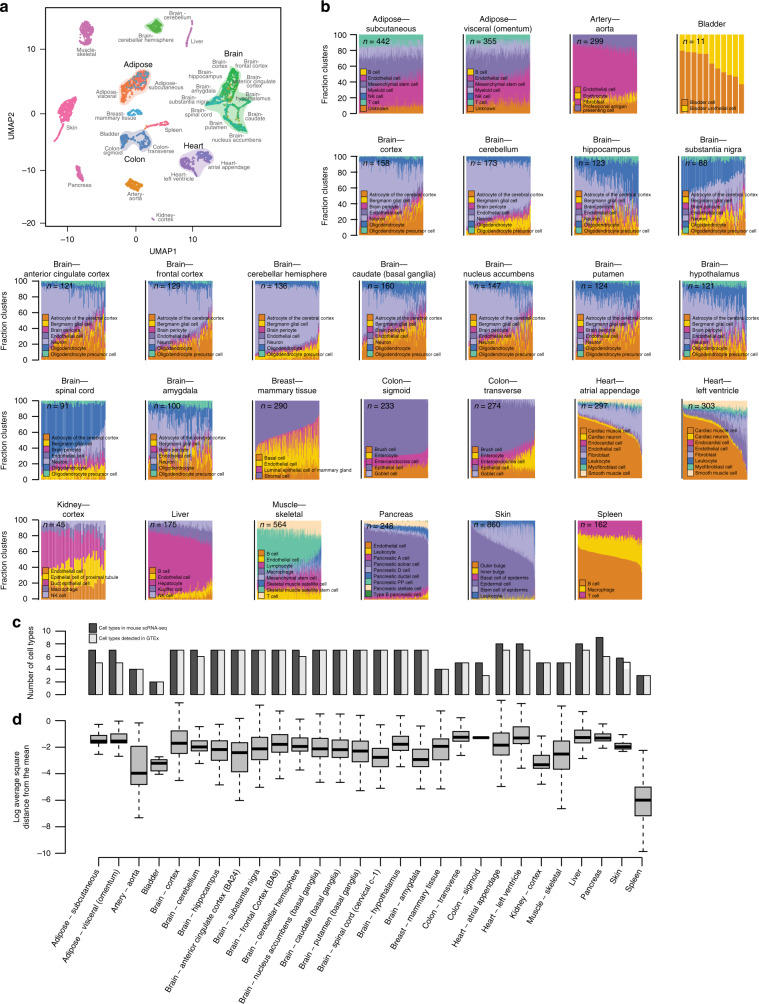
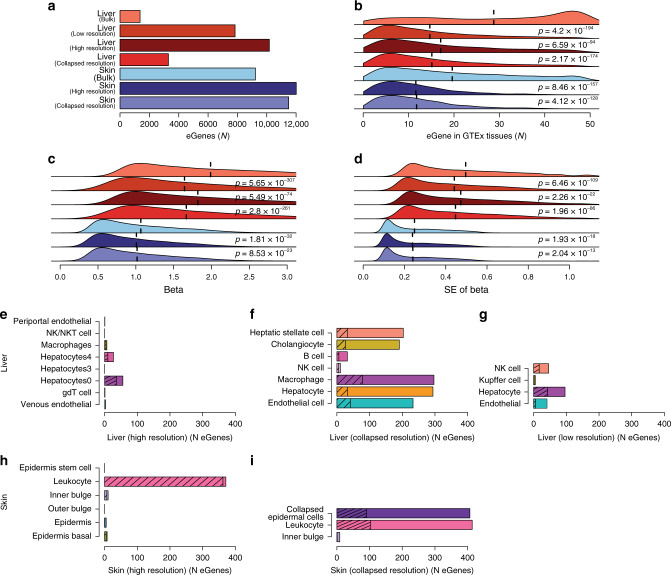
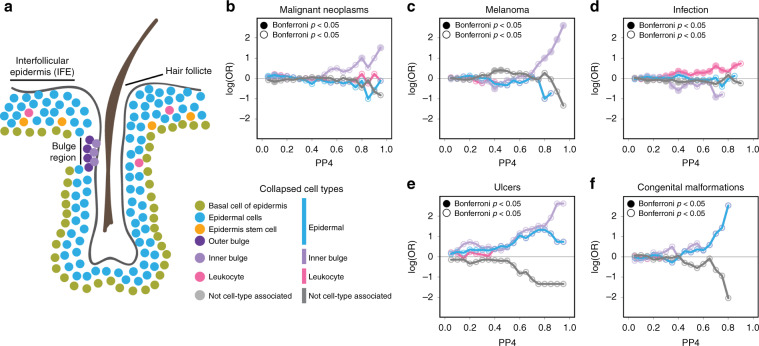
The Genotype-Tissue Expression (GTEx) resource has provided insights into the regulatory impact of genetic variation on gene expression across human tissues; however, thus far has not considered how variation acts at the resolution of the different cell types. Here, using gene expression signatures obtained from mouse cell types, we deconvolute bulk RNA-seq samples from 28 GTEx tissues to quantify cellular composition, which reveals striking heterogeneity across these samples. Conducting eQTL analyses for GTEx liver and skin samples using cell composition estimates as interaction terms, we identify thousands of genetic associations that are cell-type-associated. The skin cell-type associated eQTLs colocalize with skin diseases, indicating that variants which influence gene expression in distinct skin cell types play important roles in traits and disease. Our study provides a framework to estimate the cellular composition of GTEx tissues enabling the functional characterization of human genetic variation that impacts gene expression in cell-type-specific manners.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
