

Predicting the Landscape of Recombination Using Deep Learning
- PMID: 32077950
- PMCID: PMC7253213
- DOI: 10.1093/molbev/msaa038
Predicting the Landscape of Recombination Using Deep Learning
Abstract
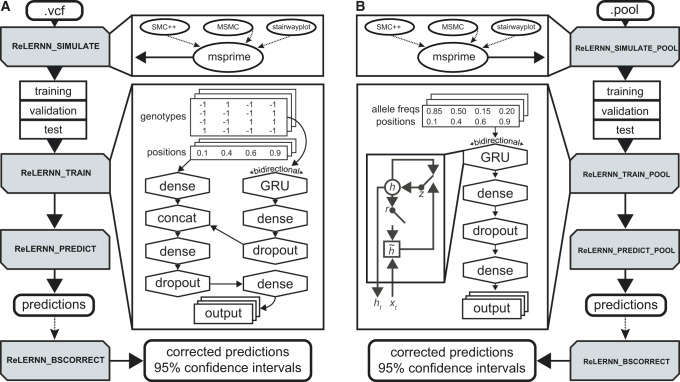
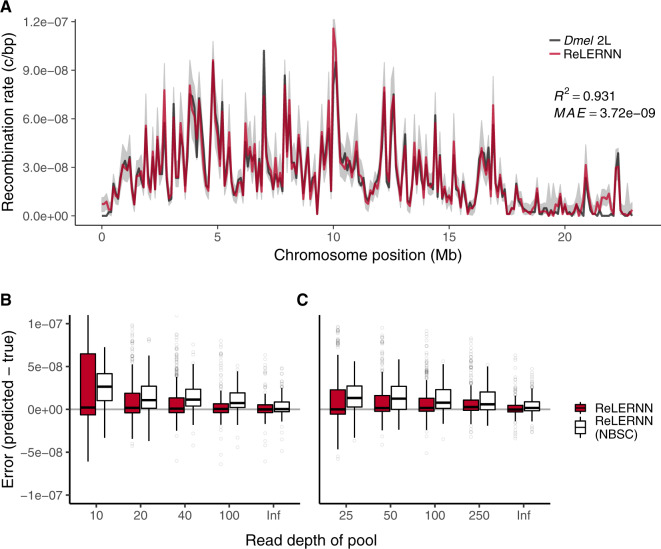
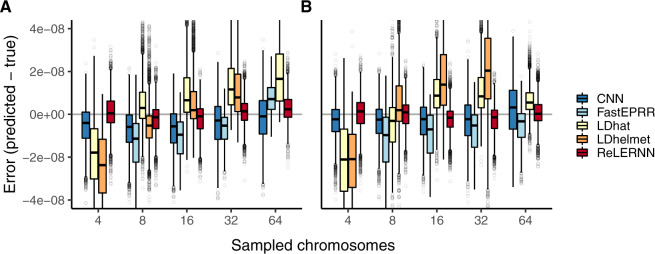
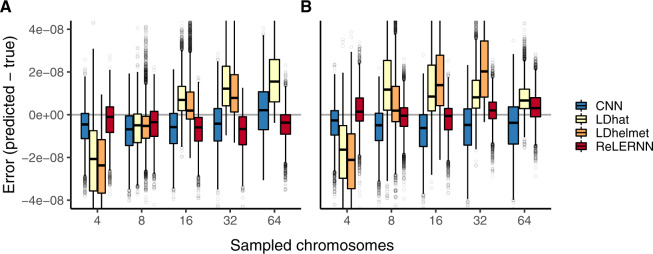
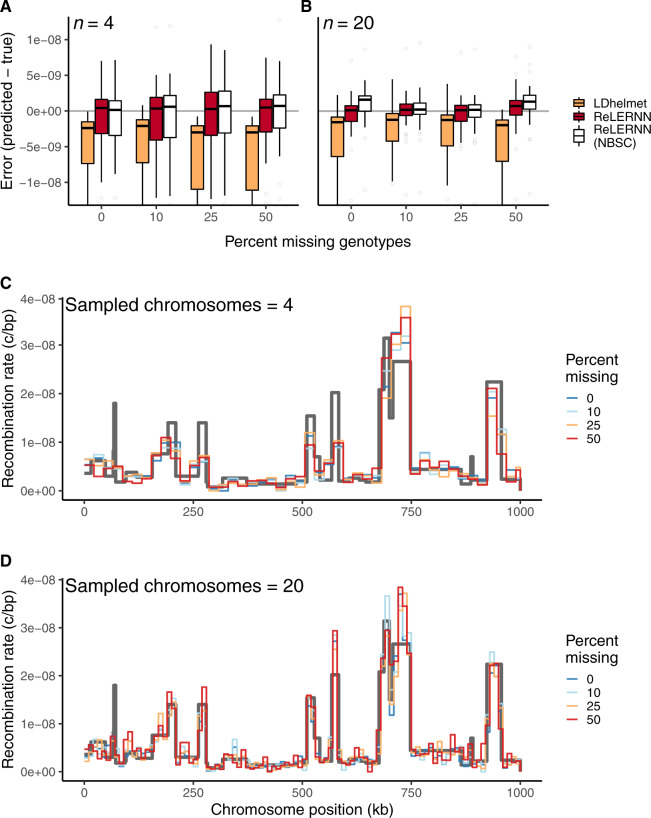
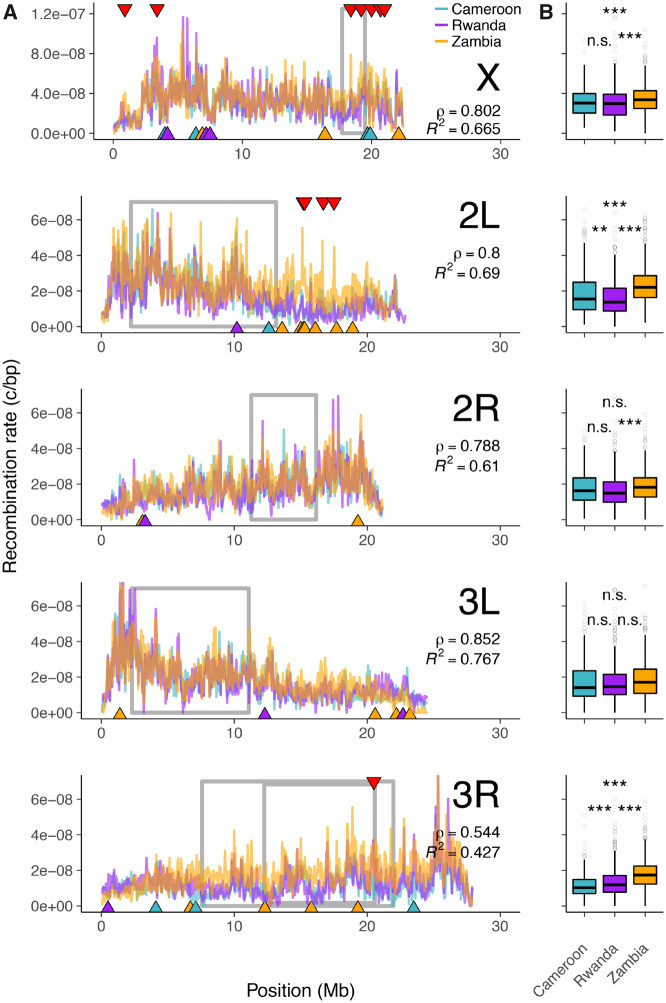
Accurately inferring the genome-wide landscape of recombination rates in natural populations is a central aim in genomics, as patterns of linkage influence everything from genetic mapping to understanding evolutionary history. Here, we describe recombination landscape estimation using recurrent neural networks (ReLERNN), a deep learning method for estimating a genome-wide recombination map that is accurate even with small numbers of pooled or individually sequenced genomes. Rather than use summaries of linkage disequilibrium as its input, ReLERNN takes columns from a genotype alignment, which are then modeled as a sequence across the genome using a recurrent neural network. We demonstrate that ReLERNN improves accuracy and reduces bias relative to existing methods and maintains high accuracy in the face of demographic model misspecification, missing genotype calls, and genome inaccessibility. We apply ReLERNN to natural populations of African Drosophila melanogaster and show that genome-wide recombination landscapes, although largely correlated among populations, exhibit important population-specific differences. Lastly, we connect the inferred patterns of recombination with the frequencies of major inversions segregating in natural Drosophila populations.
Keywords: deep learning; machine learning; population genomics; recombination.
© The Author(s) 2020. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, et al. 2015. TensorFlow: large-scale machine learning on heterogeneous systems. Available from: https://www.tensorflow.org/, software available from tensorflow.org.
-
- Aulard S, David JR, Lemeunier F.. 2002. Chromosomal inversion polymorphism in Afrotropical populations of Drosophila melanogaster. Genet Res. 79(1):49–63. - PubMed
-
- Ayala D, Guerrero RF, Kirkpatrick M.. 2013. Reproductive isolation and local adaptation quantified for a chromosome inversion in a malaria mosquito. Evolution 67(4):946–958. - PubMed
-
- Barton N. 1995. A general model for the evolution of recombination. Genet Res. 65(2):123–144. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
