

Autophagy interferes with human cytomegalovirus genome replication, morphogenesis, and progeny release
- PMID: 32079454
- PMCID: PMC8032242
- DOI: 10.1080/15548627.2020.1732686
Autophagy interferes with human cytomegalovirus genome replication, morphogenesis, and progeny release
Abstract
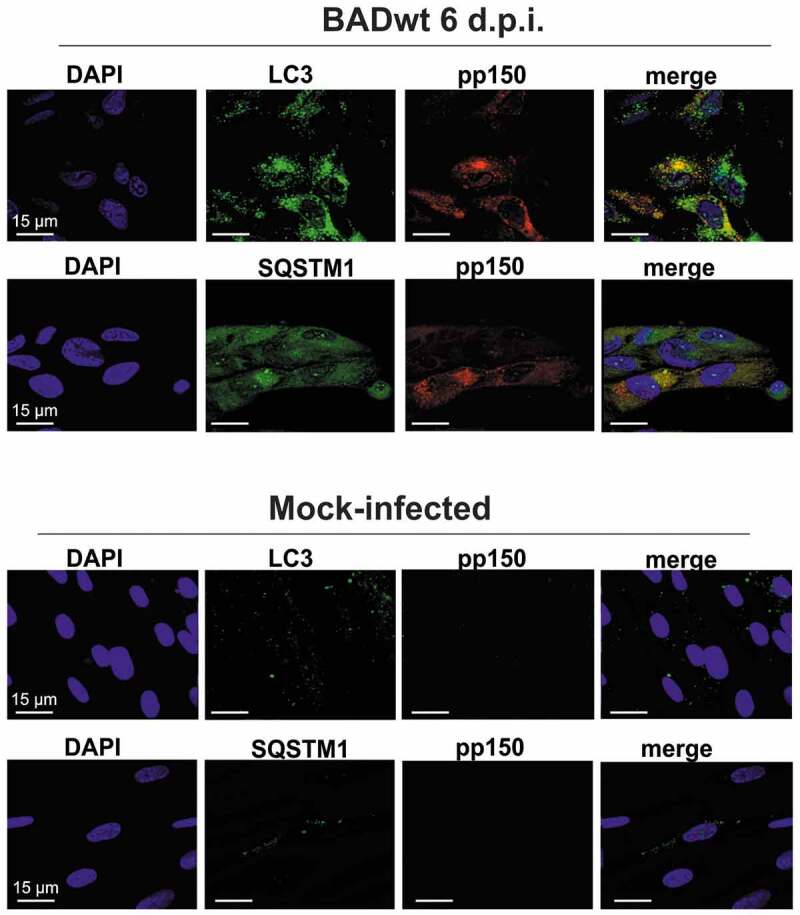
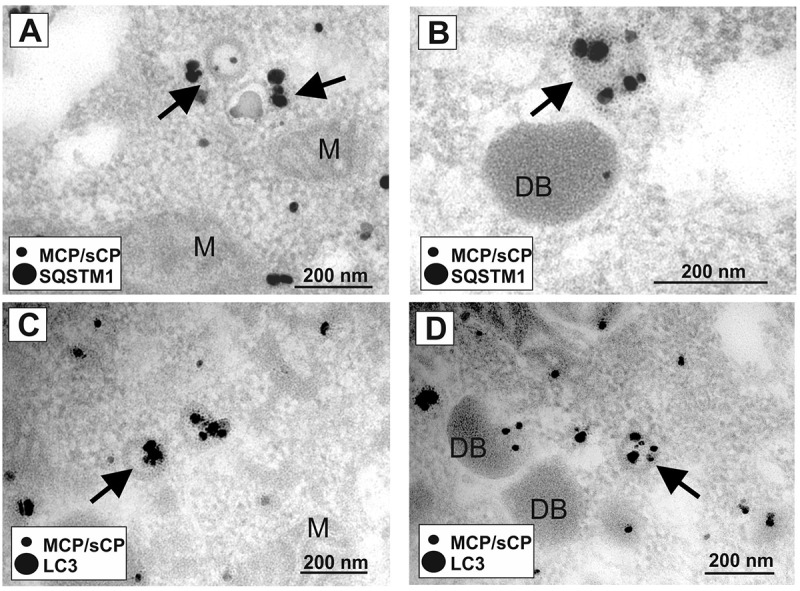
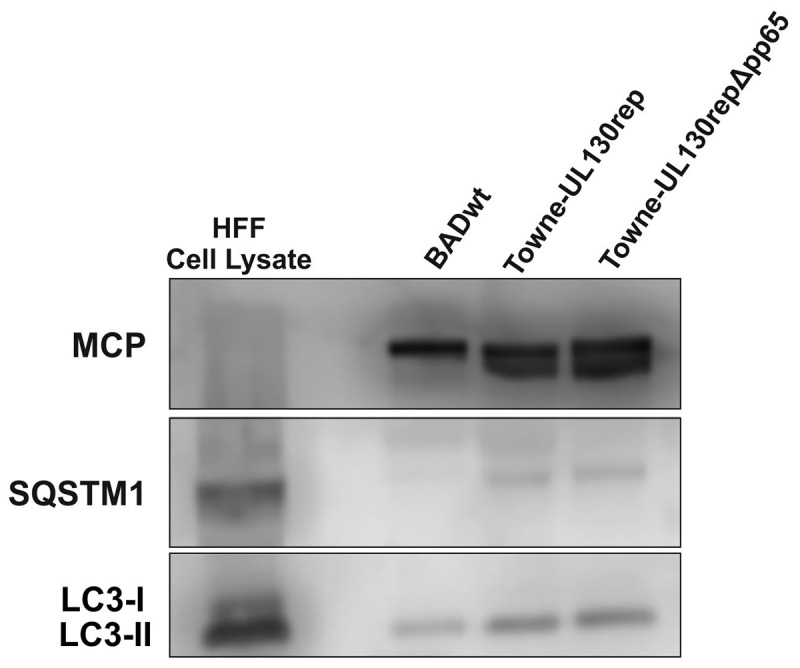
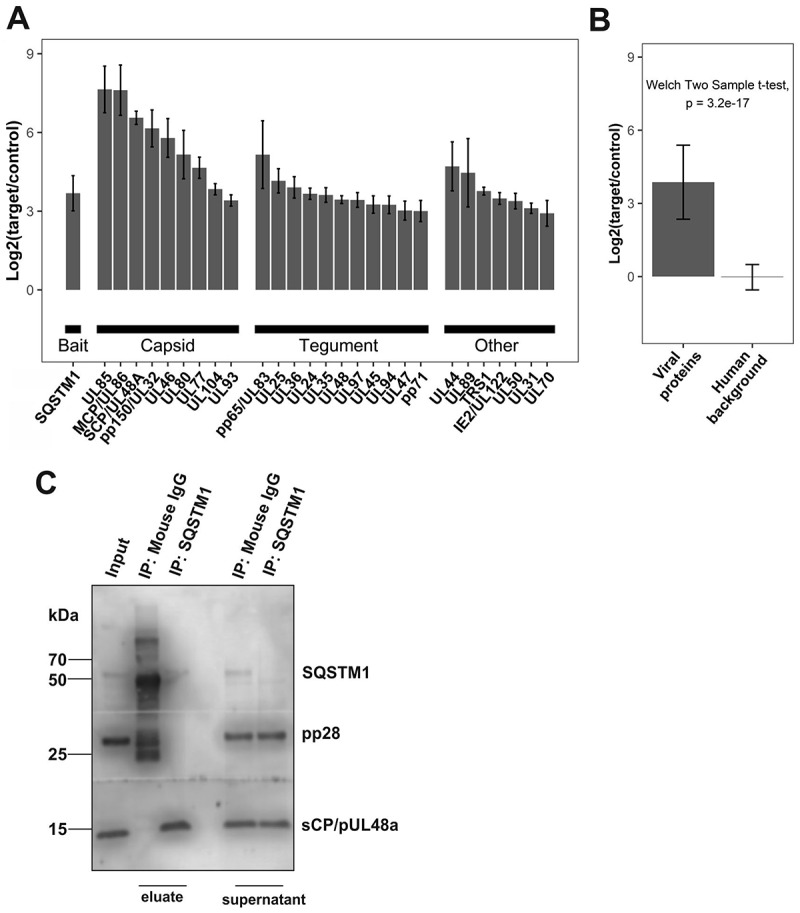
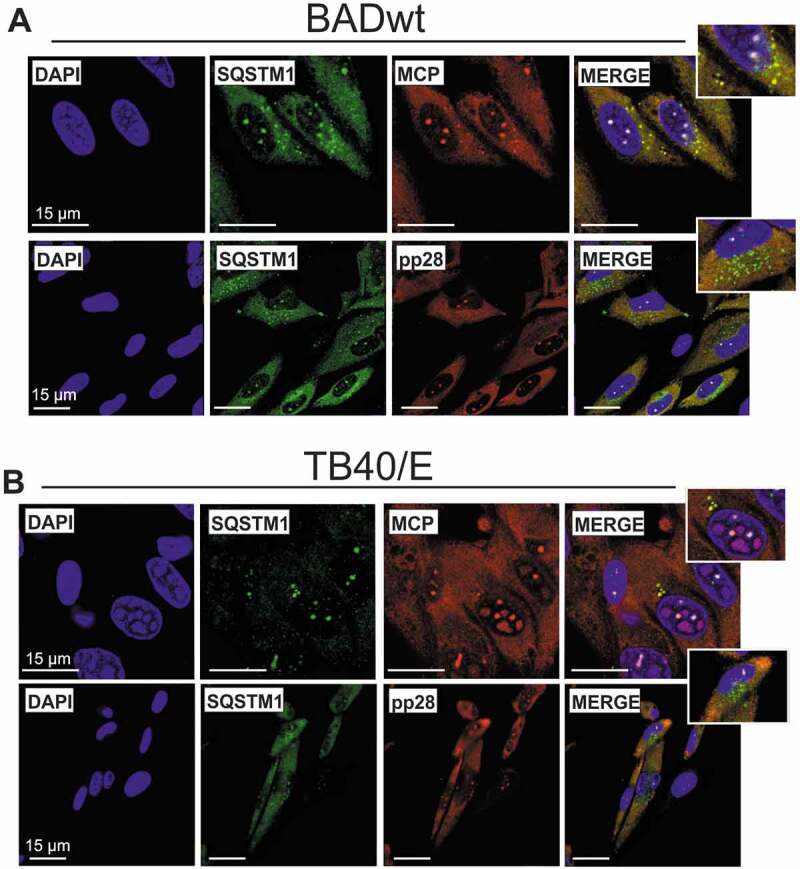
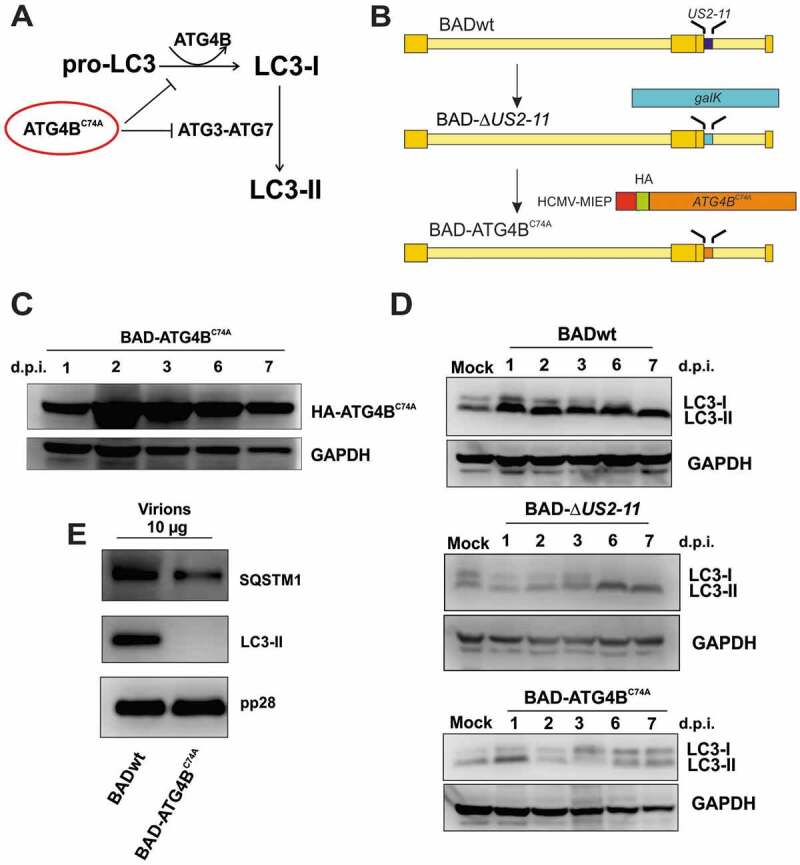
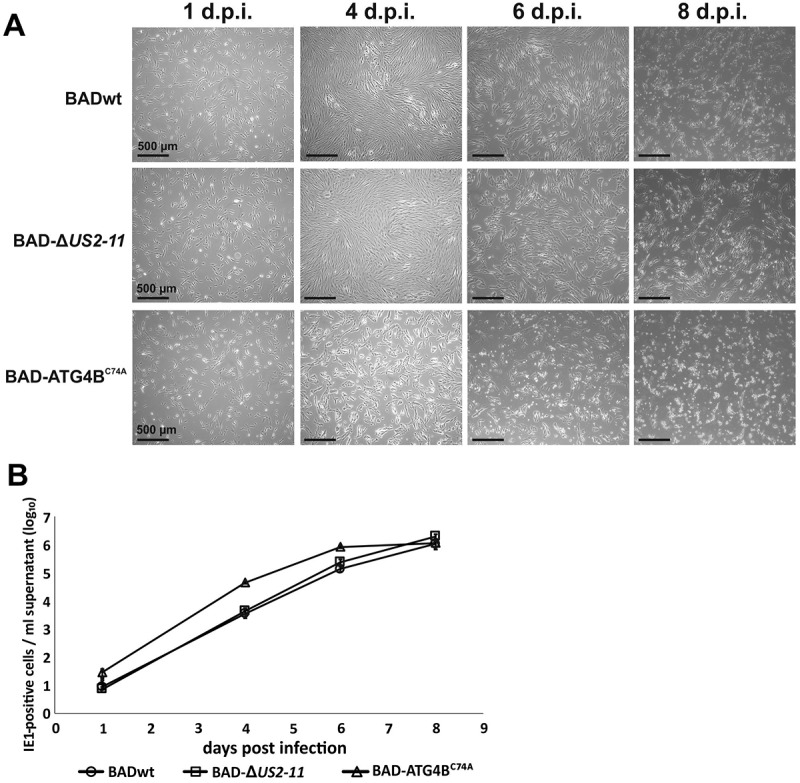
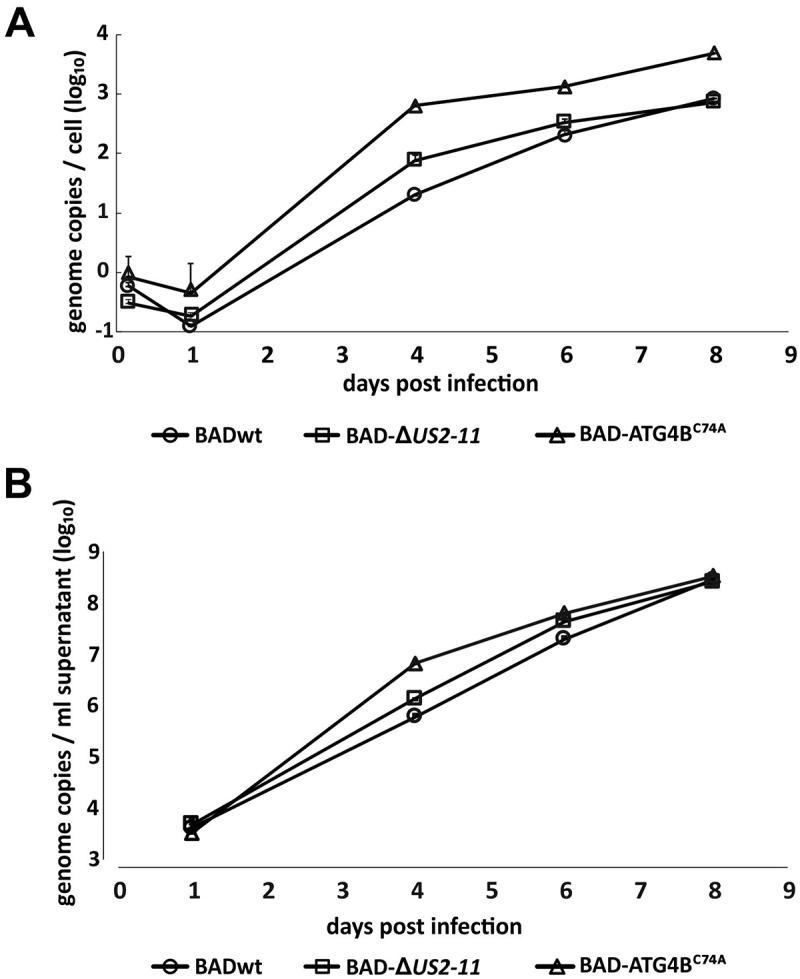
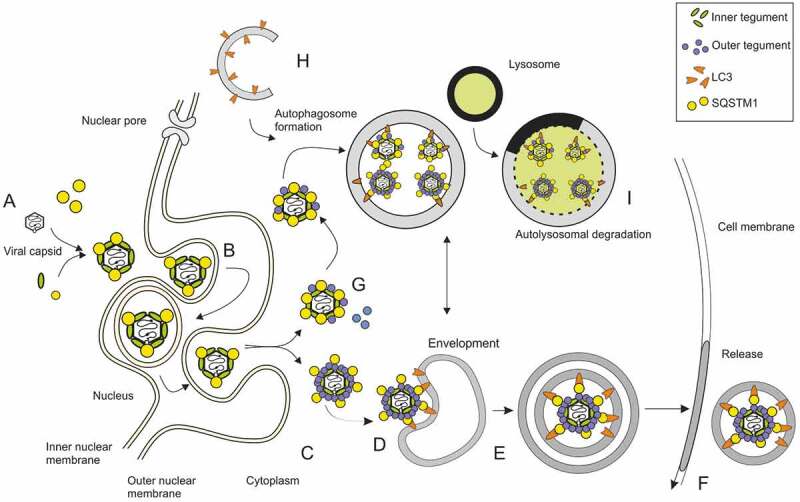
Viral infections are often accompanied by the induction of autophagy as an intrinsic cellular defense mechanism. Herpesviruses have developed strategies to evade autophagic degradation and to manipulate autophagy of the host cells to their benefit. Here we addressed the role of macroautophagy/autophagy in human cytomegalovirus replication and for particle morphogenesis. We found that proteins of the autophagy machinery localize to cytoplasmic viral assembly compartments and enveloped virions in the cytoplasm. Surprisingly, the autophagy receptor SQSTM1/p62 was also found to colocalize with HCMV capsids in the nucleus of infected cells. This finding indicates that the autophagy machinery interacts with HCMV already at the early nuclear stages of particle morphogenesis. The membrane-bound form of LC3 and several autophagy receptors were packaged into extracellular HCMV virions. This suggested that autophagic membranes were included during secondary envelopment of HCMV virions. To further address the importance of autophagy in HCMV infection, we generated an HCMV mutant that expressed a dominant-negative version of the protease ATG4B (BAD-ATG4BC74A). The proteolytic activity of ATG4B is required for LC3 cleavage, priming it for membrane conjugation. Surprisingly, both genome replication and virus release were enhanced in cells infected with BAD-ATG4BC74A, compared to control strains. These results show that autophagy operates as an antiviral process during HCMV infection but is dispensable for secondary HCMV particle envelopment.Abbreviations: ATG: autophagy-related; BAC: bacterial artificial chromosome; BECN1: beclin 1; CPE: cytopathic effect; cVACs: cytoplasmic viral assembly compartments; d.p.i.: days post-infection; DB: dense body; EBV: Epstein-Barr virus; galK: galactokinase; HCMV: human cytomegalovirus; HFF: human foreskin fibroblasts; IE: immediate-early; IRS: internal repeat short; LC3: MAP1LC3A/B; m.o.i.; multiplicity of infection; MCP: major capsid protein; Pp: phosphoprotein; sCP/UL48a: smallest capsid protein; TRS: terminal repeat short; UL: unique long; US: unique short.
Keywords: Cellular host defense; herpesviruses; human cytomegalovirus; viral morphogenesis; xenophagy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Xie Z, Klionsky DJ.. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109. - PubMed
-
- Munz C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol Rev. 2016;272(1):17–27. - PubMed
-
- Munz C. The macroautophagy machinery in endo- and exocytosis. J Mol Biol. 2017;429(4):473–485. - PubMed
-
- Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous