

Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence
- PMID: 32080423
- PMCID: PMC7849055
- DOI: 10.1038/s41569-020-0339-2
Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence
Abstract
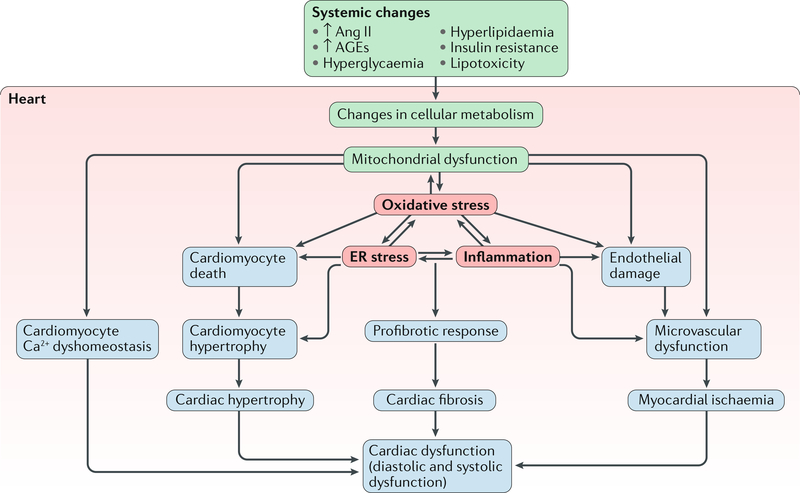
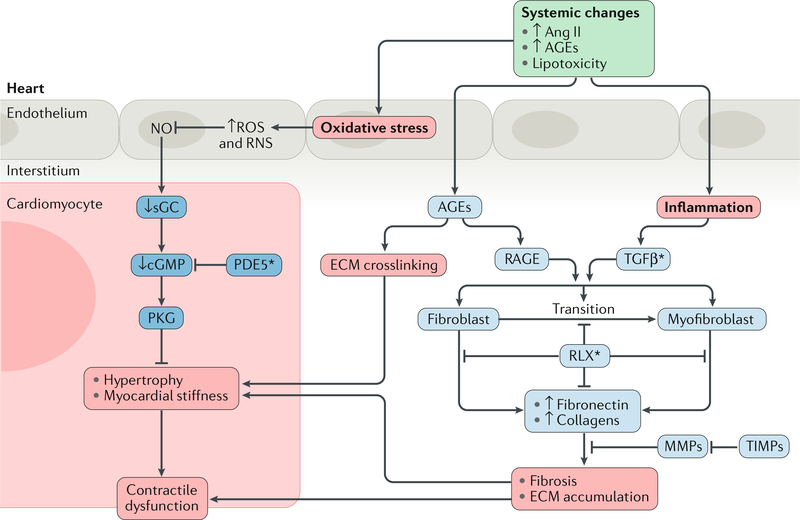
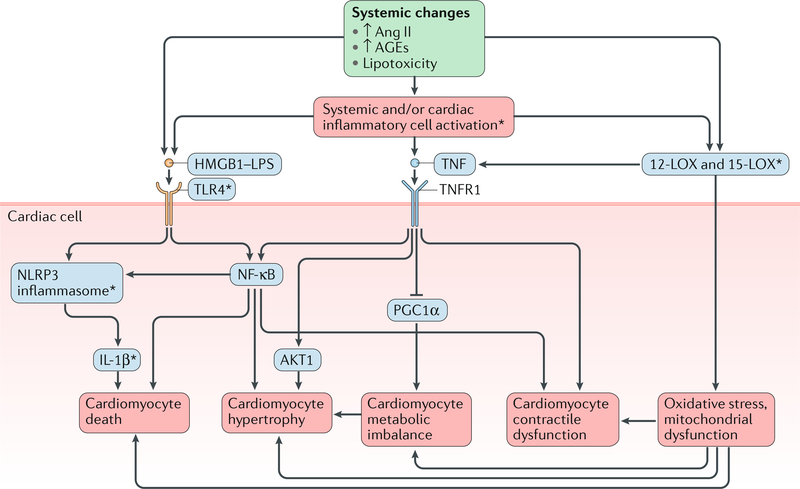
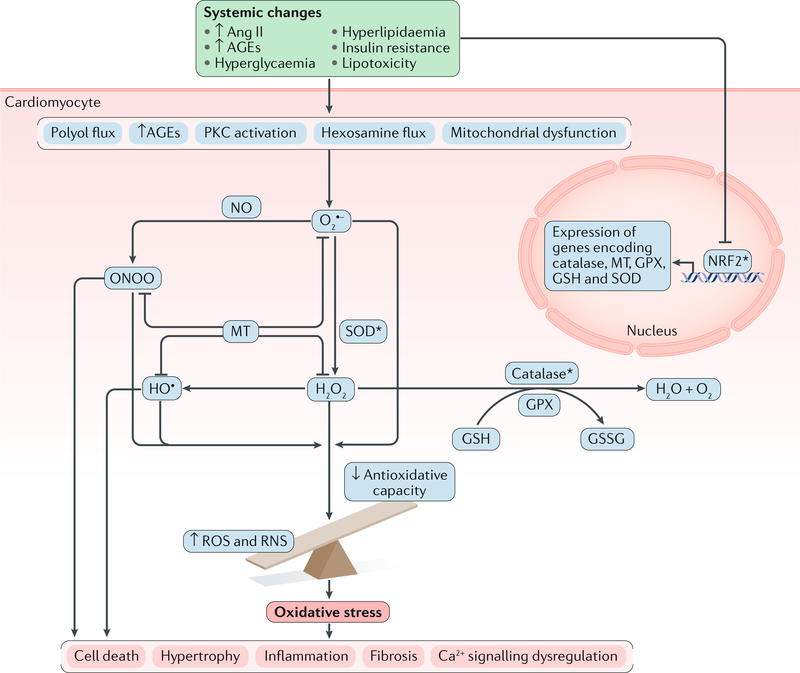
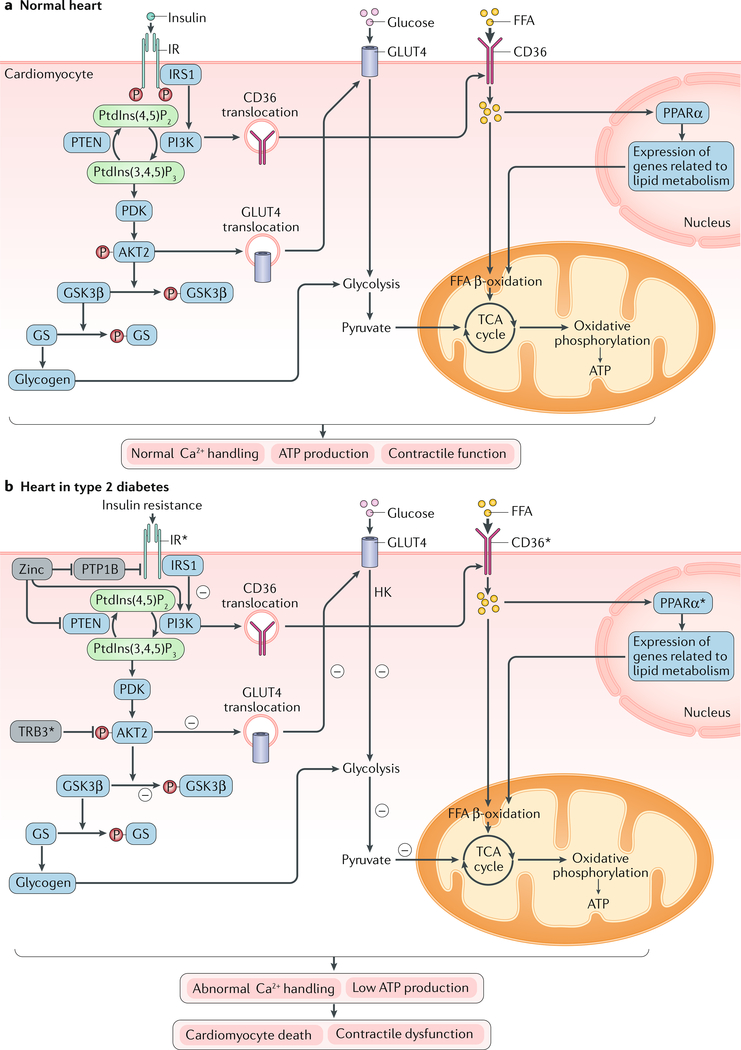
The pathogenesis and clinical features of diabetic cardiomyopathy have been well-studied in the past decade, but effective approaches to prevent and treat this disease are limited. Diabetic cardiomyopathy occurs as a result of the dysregulated glucose and lipid metabolism associated with diabetes mellitus, which leads to increased oxidative stress and the activation of multiple inflammatory pathways that mediate cellular and extracellular injury, pathological cardiac remodelling, and diastolic and systolic dysfunction. Preclinical studies in animal models of diabetes have identified multiple intracellular pathways involved in the pathogenesis of diabetic cardiomyopathy and potential cardioprotective strategies to prevent and treat the disease, including antifibrotic agents, anti-inflammatory agents and antioxidants. Some of these interventions have been tested in clinical trials and have shown favourable initial results. In this Review, we discuss the mechanisms underlying the development of diabetic cardiomyopathy and heart failure in type 1 and type 2 diabetes mellitus, and we summarize the evidence from preclinical and clinical studies that might provide guidance for the development of targeted strategies. We also highlight some of the novel pharmacological therapeutic strategies for the treatment and prevention of diabetic cardiomyopathy.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Rubler S. et al. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 30, 595–602 (1972). - PubMed
-
- Kannel WB, Hjortland M & Castelli WP Role of diabetes in congestive heart failure: the Framingham study. Am. J. Cardiol. 34, 29–34 (1974). - PubMed
-
- Tarquini R et al. Clinical approach to diabetic cardiomyopathy: a review of human studies. Curr. Med. Chem. 25, 1510–1524 (2018). - PubMed
-
- Dauriz M et al. Prognostic impact of diabetes on long-term survival outcomes in patients with heart failure: a meta-analysis. Diabetes Care 40, 1597–1605 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
