

Developments in the treatment of Fabry disease
- PMID: 32083331
- PMCID: PMC7540041
- DOI: 10.1002/jimd.12228
Developments in the treatment of Fabry disease
Abstract
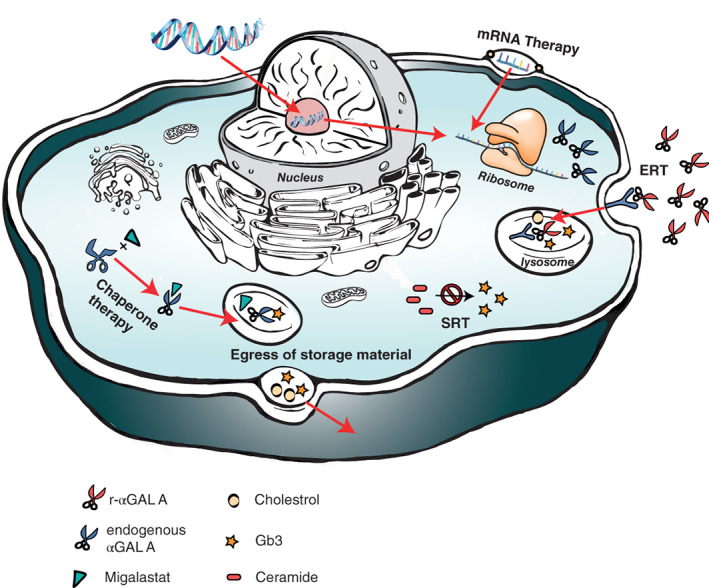
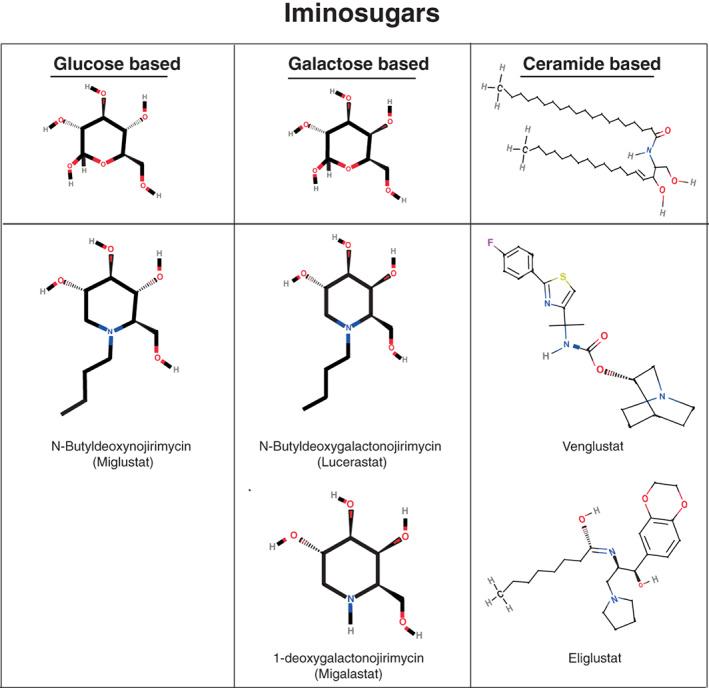
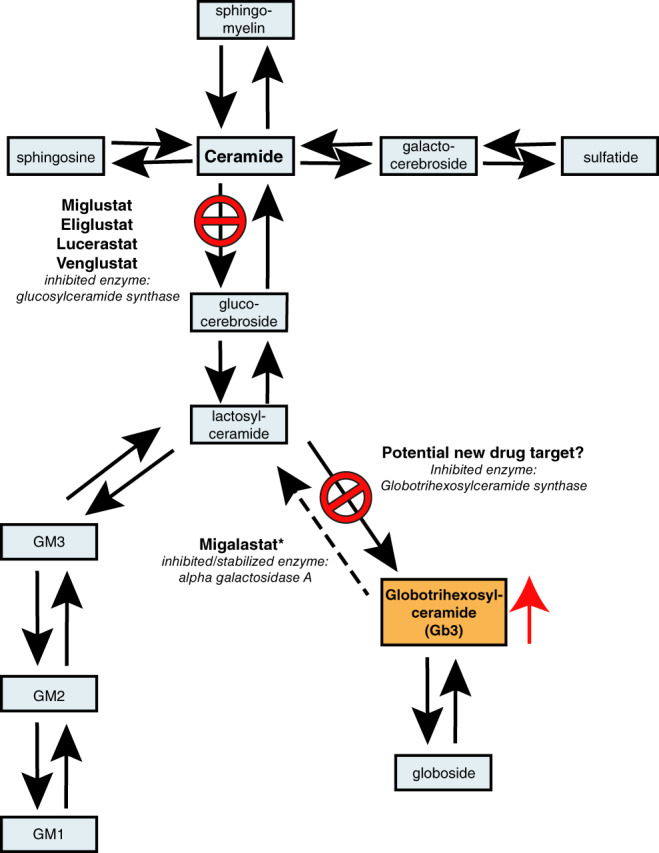
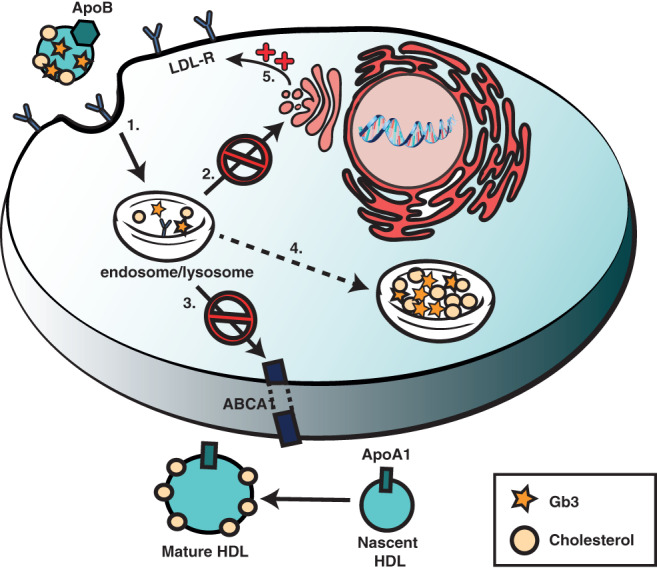
Enzyme replacement therapy (ERT) with recombinant α-galactosidase A (r-αGAL A) for the treatment of Fabry disease has been available for over 15 years. Long-term treatment may slow down disease progression, but cardiac, renal, and cerebral complications still develop in most patients. In addition, lifelong intravenous treatment is burdensome. Therefore, several new treatment approaches have been explored over the past decade. Chaperone therapy (Migalastat; 1-deoxygalactonojirimycin) is the only other currently approved therapy for Fabry disease. This oral small molecule aims to improve enzyme activity of mutated α-galactosidase A and can only be used in patients with specific mutations. Treatments currently under evaluation in (pre)clinical trials are second generation enzyme replacement therapies (Pegunigalsidase-alfa, Moss-aGal), substrate reduction therapies (Venglustat and Lucerastat), mRNA- and gene-based therapy. This review summarises the knowledge on currently available and potential future options for the treatment of Fabry disease.
Keywords: Fabry disease; chaperone therapy; enzyme replacement therapy (ERT); gene therapy; substrate reduction therapy (SRT); treatment.
© 2020 The Authors. Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
No fees, travel support or grants are obtained from Pharmaceutical Industry by any of the listed authors. M.L. reports to be involved in pre‐marketing studies with Genzyme, Protalix, and Idorsia. Financial arrangements are made through AMC Research BV. C.E.H. reports to be involved in pre‐marketing studies with Genzyme, Protalix, and Idorsia. Financial arrangements are made through AMC Research BV. S.J.V. reports to be involved in a pre‐marketing study with Protalix. Financial arrangements are made through AMC Research BV. A.B.P.K. has nothing to disclose.
Figures
References
-
- Askari H, Kaneski CR, Semino‐Mora C, et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch. 2007;4:823‐834. - PubMed
-
- Valbuena C, Leitao D, Carneiro F, Oliveira JP. Immunohistochemical diagnosis of Fabry nephropathy and localisation of globotriaosylceramide deposits in paraffin‐embedded kidney tissue sections. Virchows Arch. 2012;2:211‐221. - PubMed
-
- Desnick RJ, Ioannou YA, Eng CM. α‐Galactosidase A deficiency: Fabry disease In: Beaudet AL, Vogelstein B, Kinzler KW, et al., eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: The McGraw‐Hill Companies, Inc; 2014.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
