

Hierarchical chromatin organization detected by TADpole
- PMID: 32083658
- PMCID: PMC7144900
- DOI: 10.1093/nar/gkaa087
Hierarchical chromatin organization detected by TADpole
Abstract
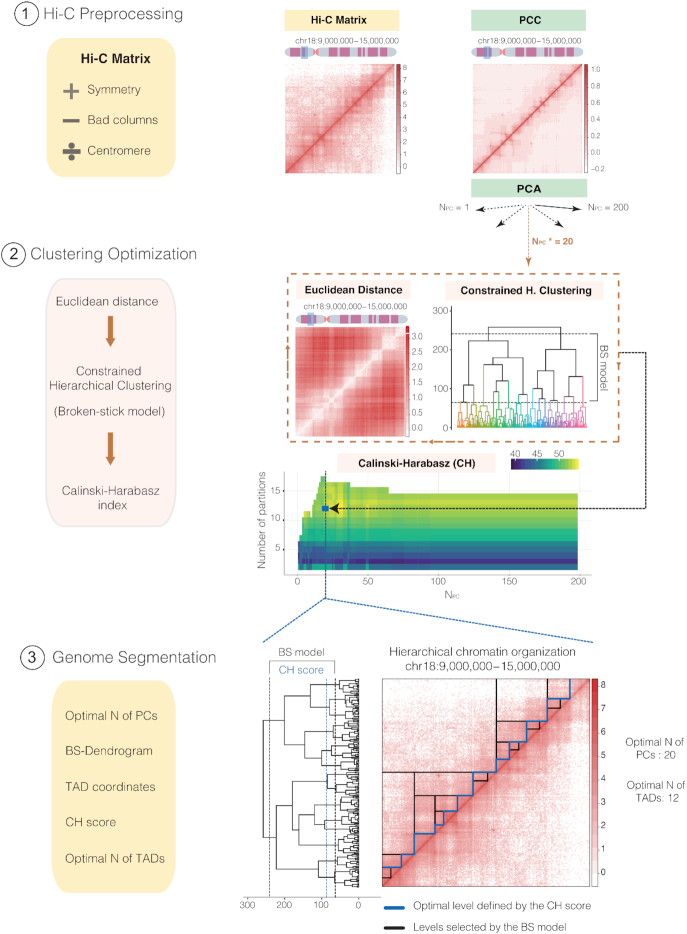
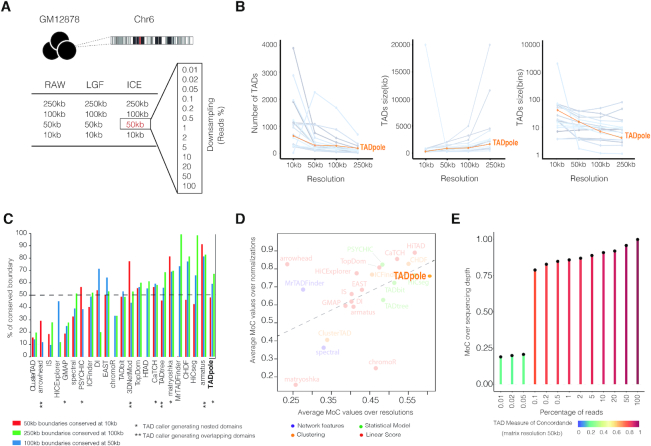
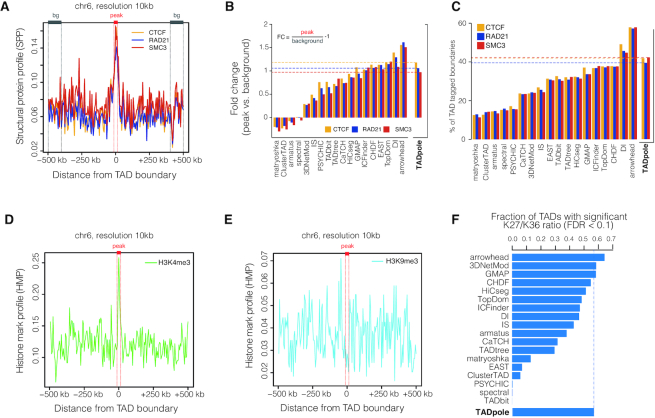
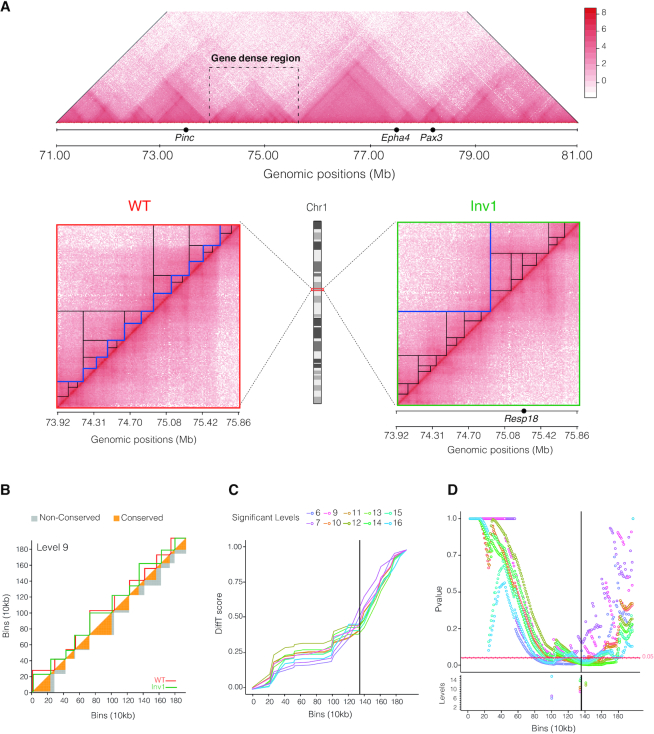
The rapid development of Chromosome Conformation Capture (3C-based techniques), as well as imaging together with bioinformatics analyses, has been fundamental for unveiling that chromosomes are organized into the so-called topologically associating domains or TADs. While TADs appear as nested patterns in the 3C-based interaction matrices, the vast majority of available TAD callers are based on the hypothesis that TADs are individual and unrelated chromatin structures. Here we introduce TADpole, a computational tool designed to identify and analyze the entire hierarchy of TADs in intra-chromosomal interaction matrices. TADpole combines principal component analysis and constrained hierarchical clustering to provide a set of significant hierarchical chromatin levels in a genomic region of interest. TADpole is robust to data resolution, normalization strategy and sequencing depth. Domain borders defined by TADpole are enriched in main architectural proteins (CTCF and cohesin complex subunits) and in the histone mark H3K4me3, while their domain bodies, depending on their activation-state, are enriched in either H3K36me3 or H3K27me3, highlighting that TADpole is able to distinguish functional TAD units. Additionally, we demonstrate that TADpole's hierarchical annotation, together with the new DiffT score, allows for detecting significant topological differences on Capture Hi-C maps between wild-type and genetically engineered mouse.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Sexton T., Cavalli G.. The role of chromosome domains in shaping the functional genome. Cell. 2015; 160:1049–1059. - PubMed
-
- Stadhouders R., Vidal E., Serra F., Di Stefano B., Le Dily F., Quilez J., Gomez A., Collombet S., Berenguer C., Cuartero Y. et al. .. Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet. 2018; 50:238–249. - PMC - PubMed
-
- Paulsen J., Liyakat Ali T.M., Nekrasov M., Delbarre E., Baudement M.O., Kurscheid S., Tremethick D., Collas P.. Long-range interactions between topologically associating domains shape the four-dimensional genome during differentiation. Nat. Genet. 2019; 51:835–843. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
