

DeepHiC: A generative adversarial network for enhancing Hi-C data resolution
- PMID: 32084131
- PMCID: PMC7055922
- DOI: 10.1371/journal.pcbi.1007287
DeepHiC: A generative adversarial network for enhancing Hi-C data resolution
Abstract
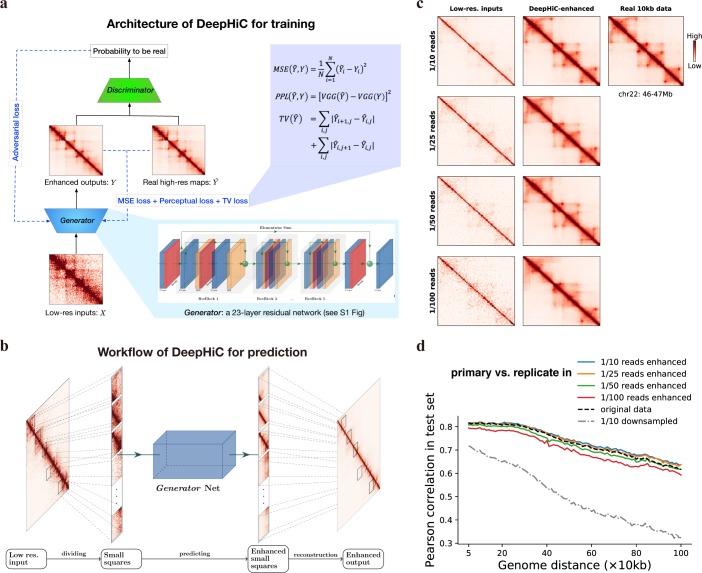
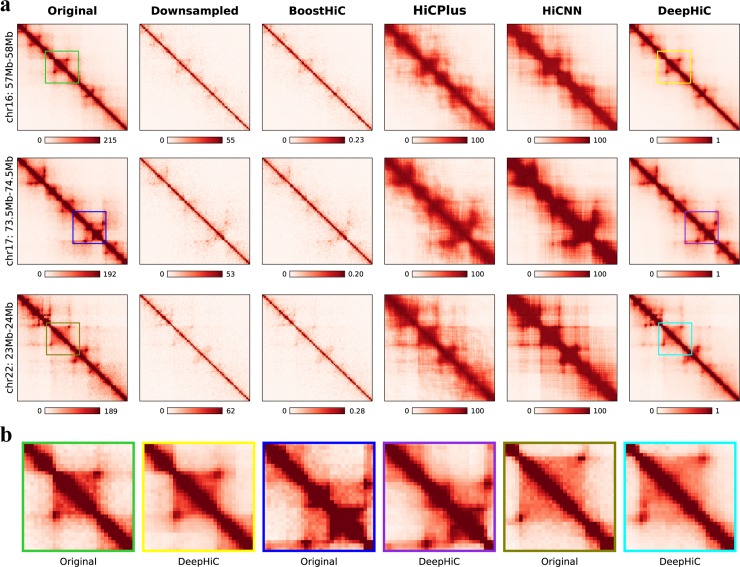
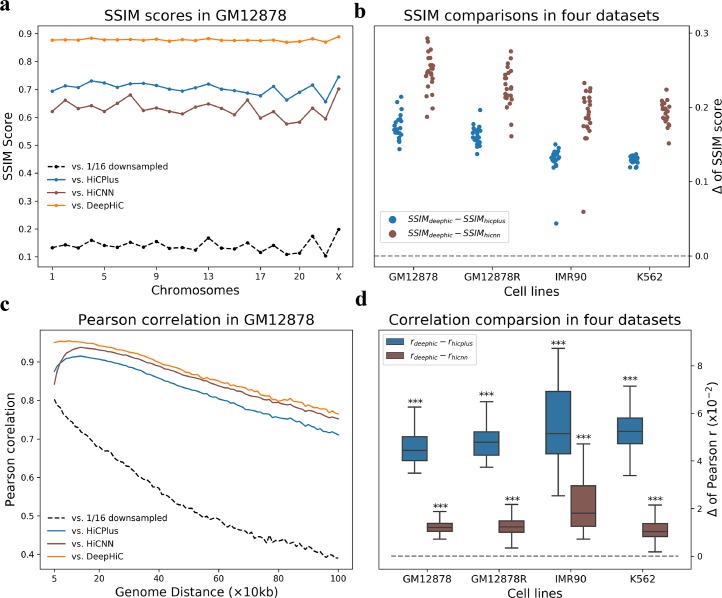
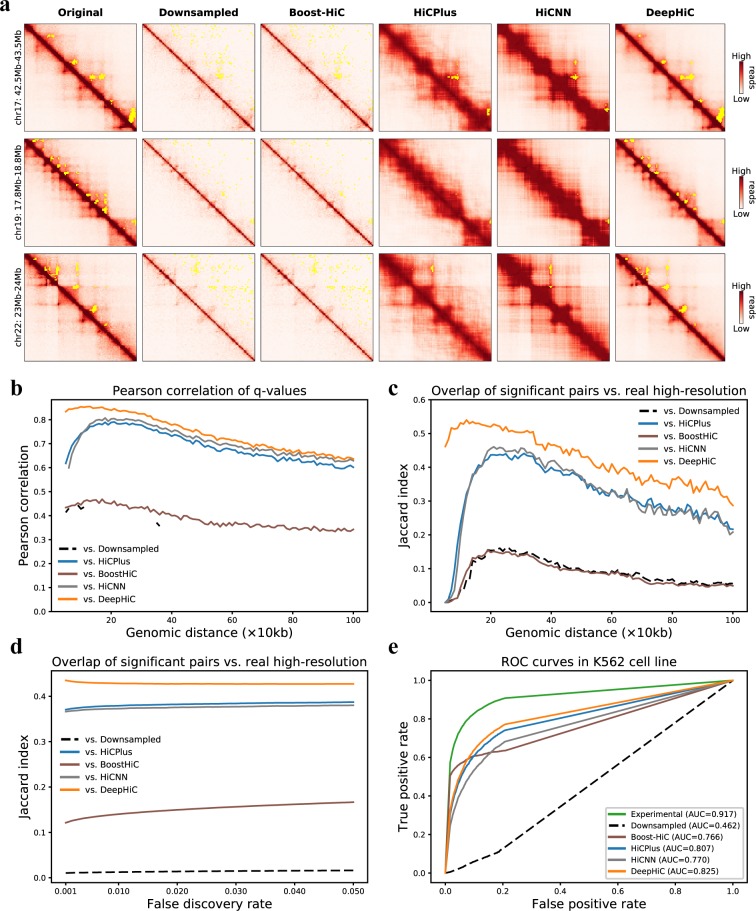
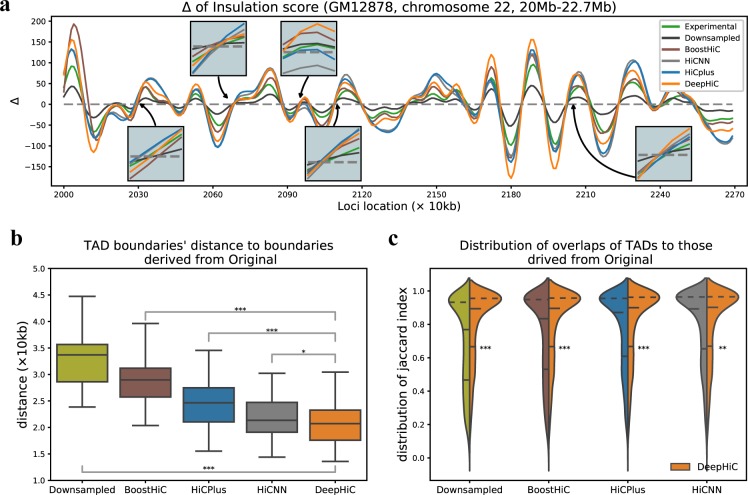
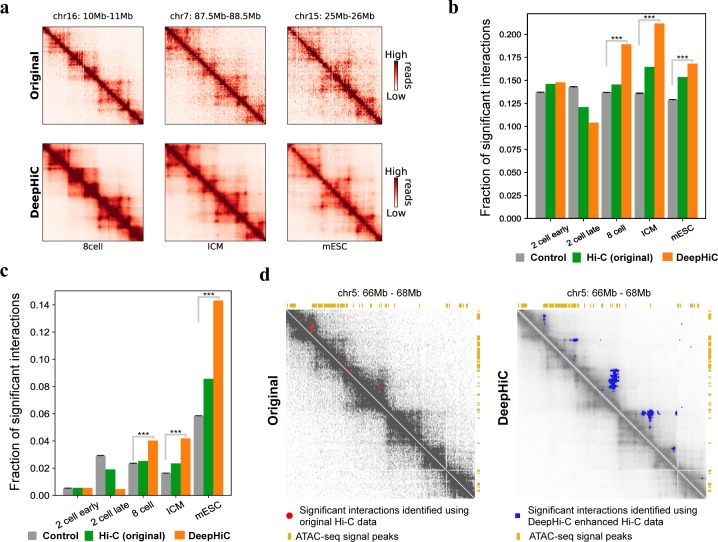
Hi-C is commonly used to study three-dimensional genome organization. However, due to the high sequencing cost and technical constraints, the resolution of most Hi-C datasets is coarse, resulting in a loss of information and biological interpretability. Here we develop DeepHiC, a generative adversarial network, to predict high-resolution Hi-C contact maps from low-coverage sequencing data. We demonstrated that DeepHiC is capable of reproducing high-resolution Hi-C data from as few as 1% downsampled reads. Empowered by adversarial training, our method can restore fine-grained details similar to those in high-resolution Hi-C matrices, boosting accuracy in chromatin loops identification and TADs detection, and outperforms the state-of-the-art methods in accuracy of prediction. Finally, application of DeepHiC to Hi-C data on mouse embryonic development can facilitate chromatin loop detection. We develop a web-based tool (DeepHiC, http://sysomics.com/deephic) that allows researchers to enhance their own Hi-C data with just a few clicks.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
