

Passenger Mutations in More Than 2,500 Cancer Genomes: Overall Molecular Functional Impact and Consequences
- PMID: 32084333
- PMCID: PMC7210002
- DOI: 10.1016/j.cell.2020.01.032
Passenger Mutations in More Than 2,500 Cancer Genomes: Overall Molecular Functional Impact and Consequences
Abstract
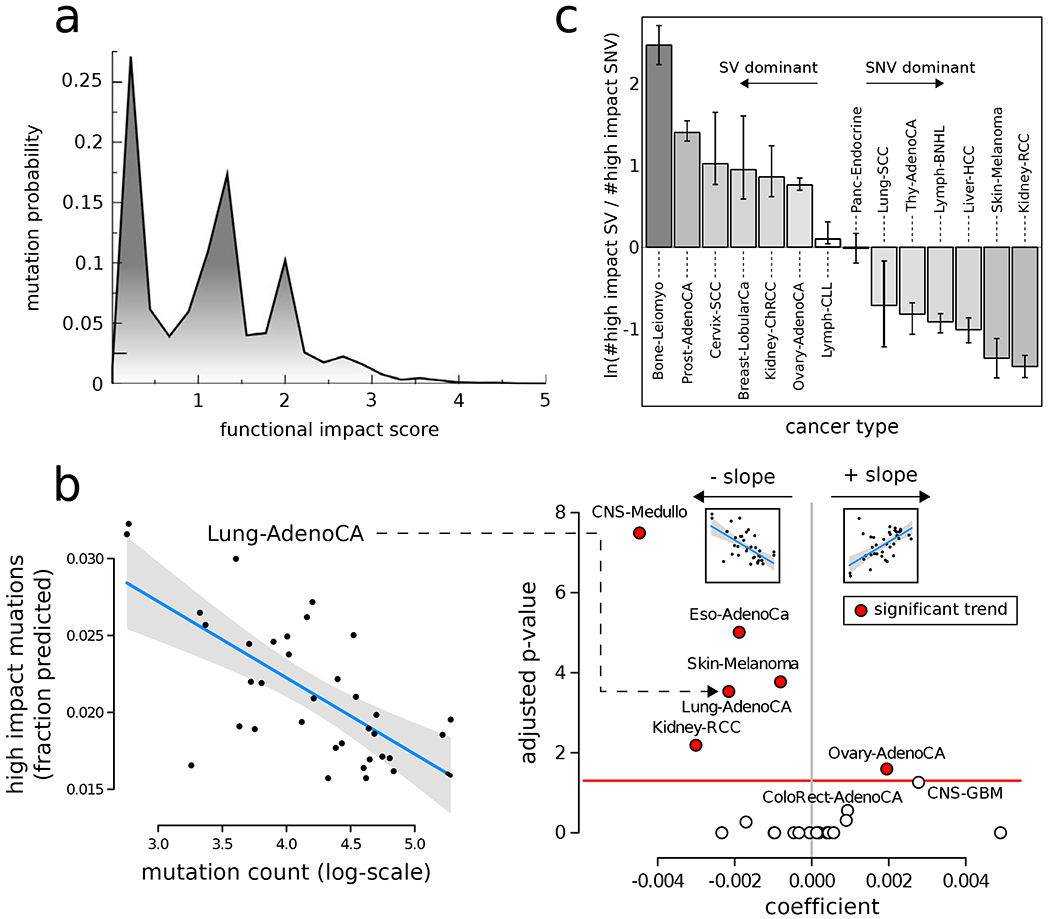
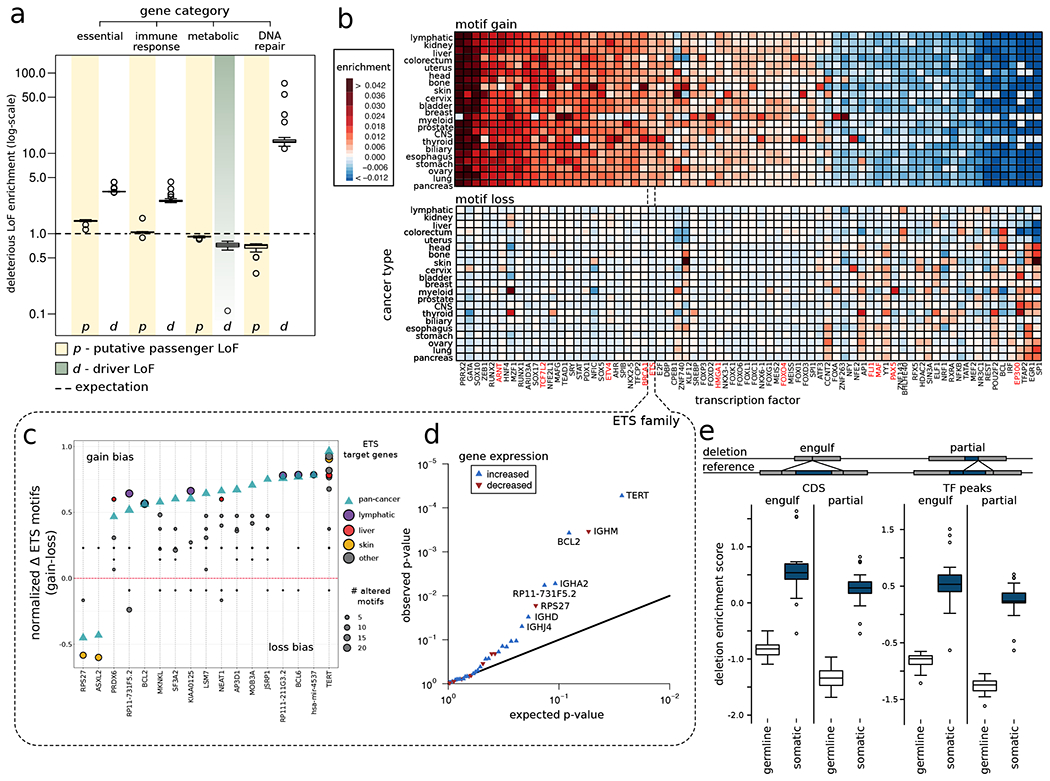
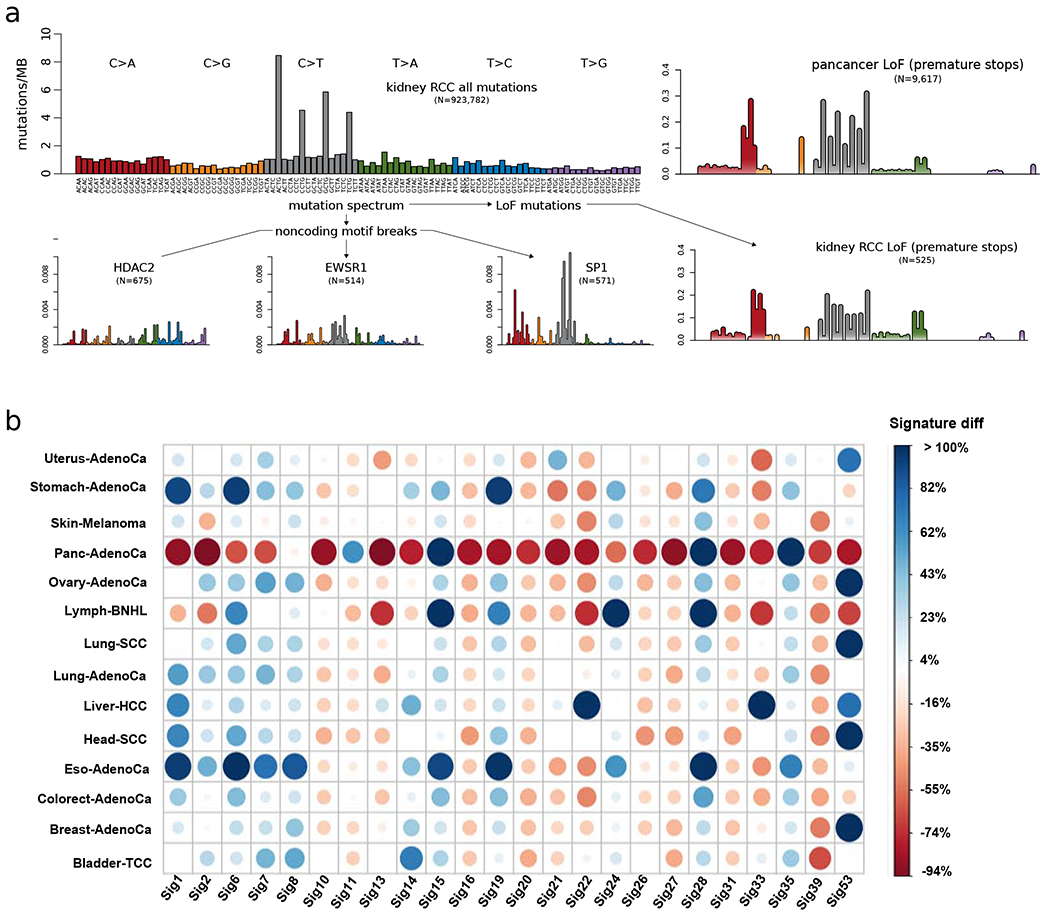
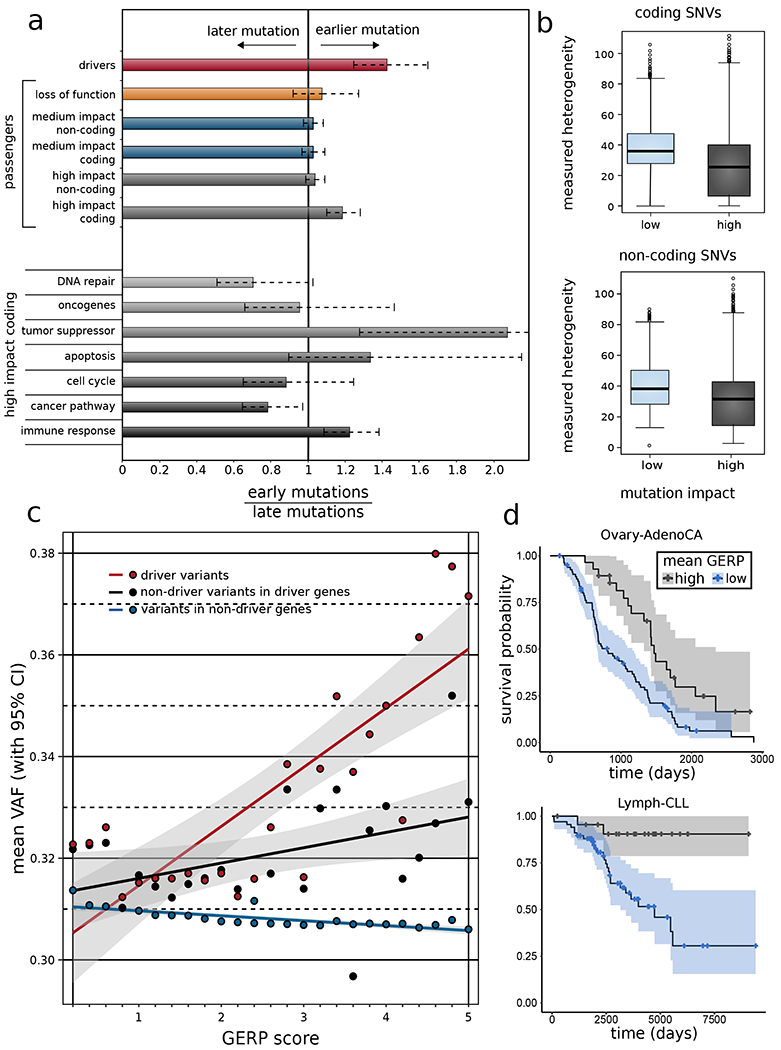
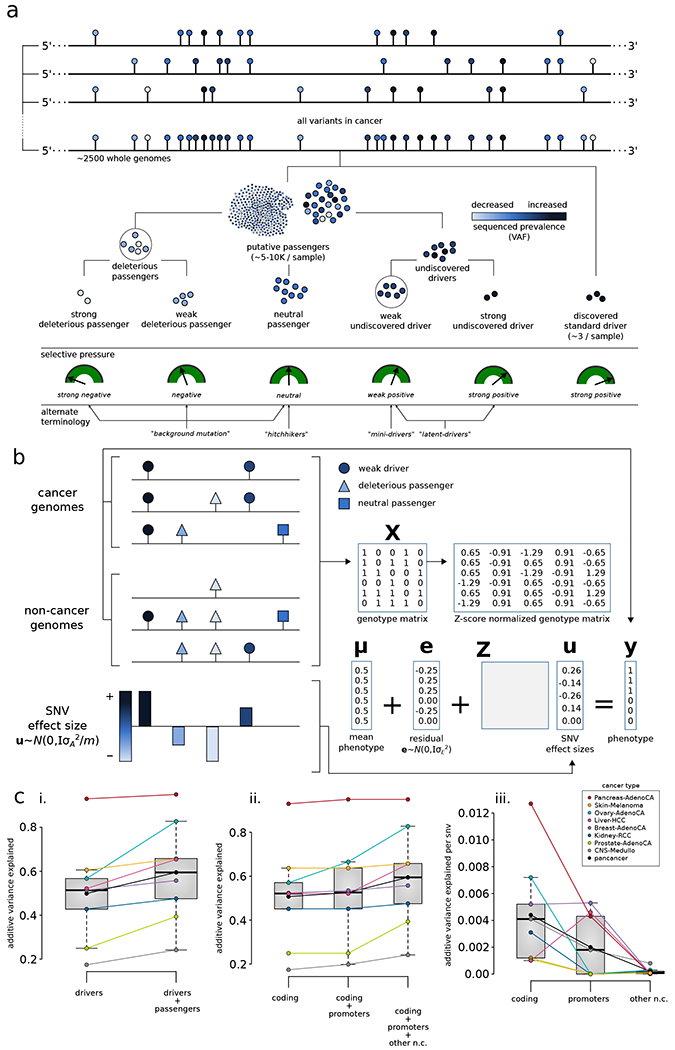
The dichotomous model of "drivers" and "passengers" in cancer posits that only a few mutations in a tumor strongly affect its progression, with the remaining ones being inconsequential. Here, we leveraged the comprehensive variant dataset from the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (PCAWG) project to demonstrate that-in addition to the dichotomy of high- and low-impact variants-there is a third group of medium-impact putative passengers. Moreover, we also found that molecular impact correlates with subclonal architecture (i.e., early versus late mutations), and different signatures encode for mutations with divergent impact. Furthermore, we adapted an additive-effects model from complex-trait studies to show that the aggregated effect of putative passengers, including undetected weak drivers, provides significant additional power (∼12% additive variance) for predicting cancerous phenotypes, beyond PCAWG-identified driver mutations. Finally, this framework allowed us to estimate the frequency of potential weak-driver mutations in PCAWG samples lacking any well-characterized driver alterations.
Keywords: PCAWG; additive-efects; cancer genomics; deleterious passengers; driver mutations; molecular impact; passenger mutations; weak drivers.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests G.G. received funds from IBM and Pharmacyclics. G.G. is listed as inventor for multiple patent applications including MuTect, ABSOLUTE, POLYSOLVER, MutSig, and MSMuTect.
Figures
References
-
- Alexandrov L, Kim J, Haradhvala NJ, Huang MN, Ng AWT, Boot A, Covington KR, Gordenin DA, Bergstrom E, Lopez-Bigas N, et al. (2018). The Repertoire of Mutational Signatures in Human Cancer. BioRxiv 322859.
-
- Campbell PJ, Getz G, Stuart JM, Korbel JO, Stein LD, and Net, - ICGC/TCGA Pan-Cancer Analysis of Whole Genomes (2017). Pan-cancer analysis of whole genomes. BioRxiv 162784.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
