

Relapse-Fated Latent Diagnosis Subclones in Acute B Lineage Leukemia Are Drug Tolerant and Possess Distinct Metabolic Programs
- PMID: 32086311
- PMCID: PMC7122013
- DOI: 10.1158/2159-8290.CD-19-1059
Relapse-Fated Latent Diagnosis Subclones in Acute B Lineage Leukemia Are Drug Tolerant and Possess Distinct Metabolic Programs
Abstract
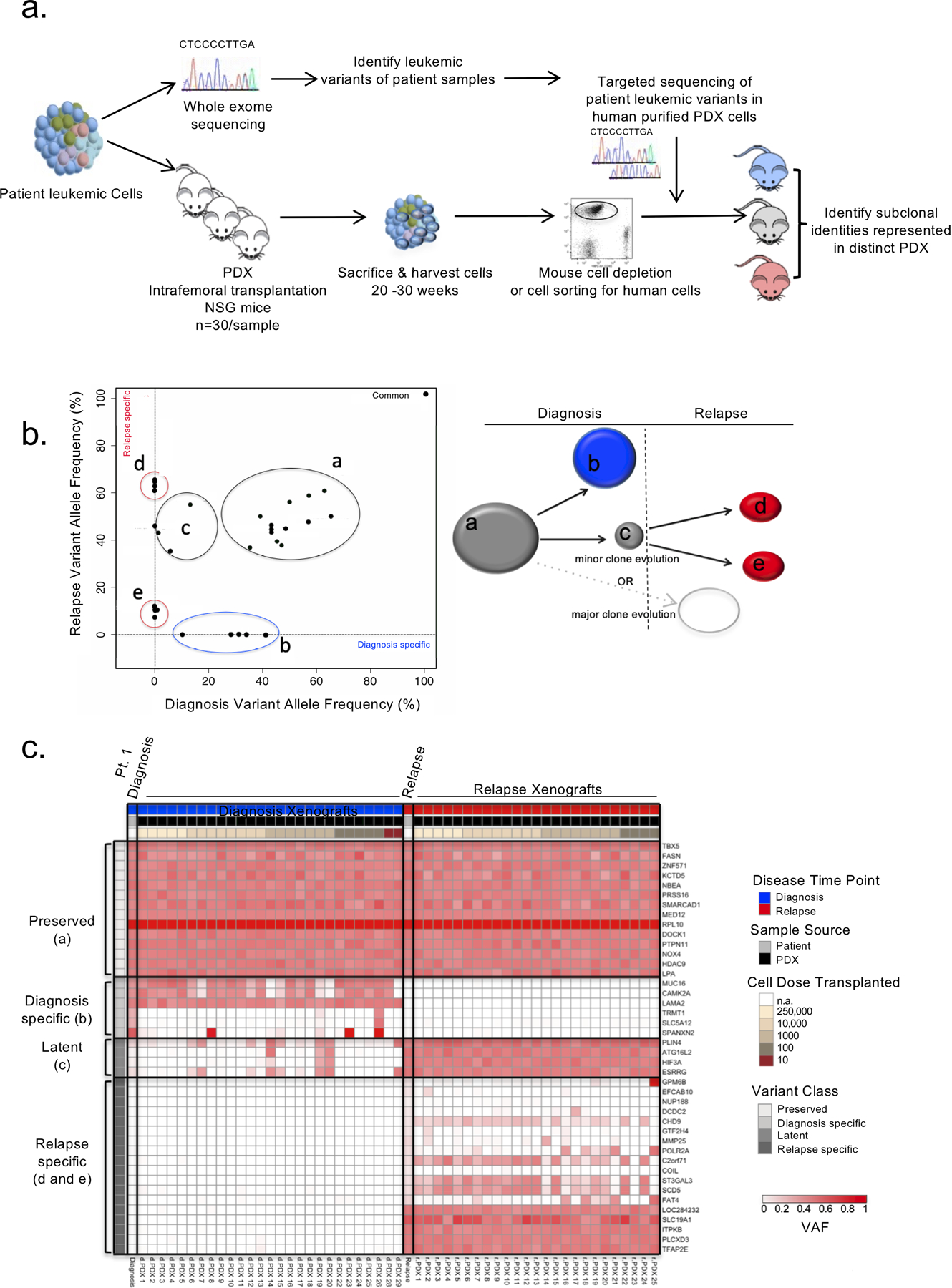
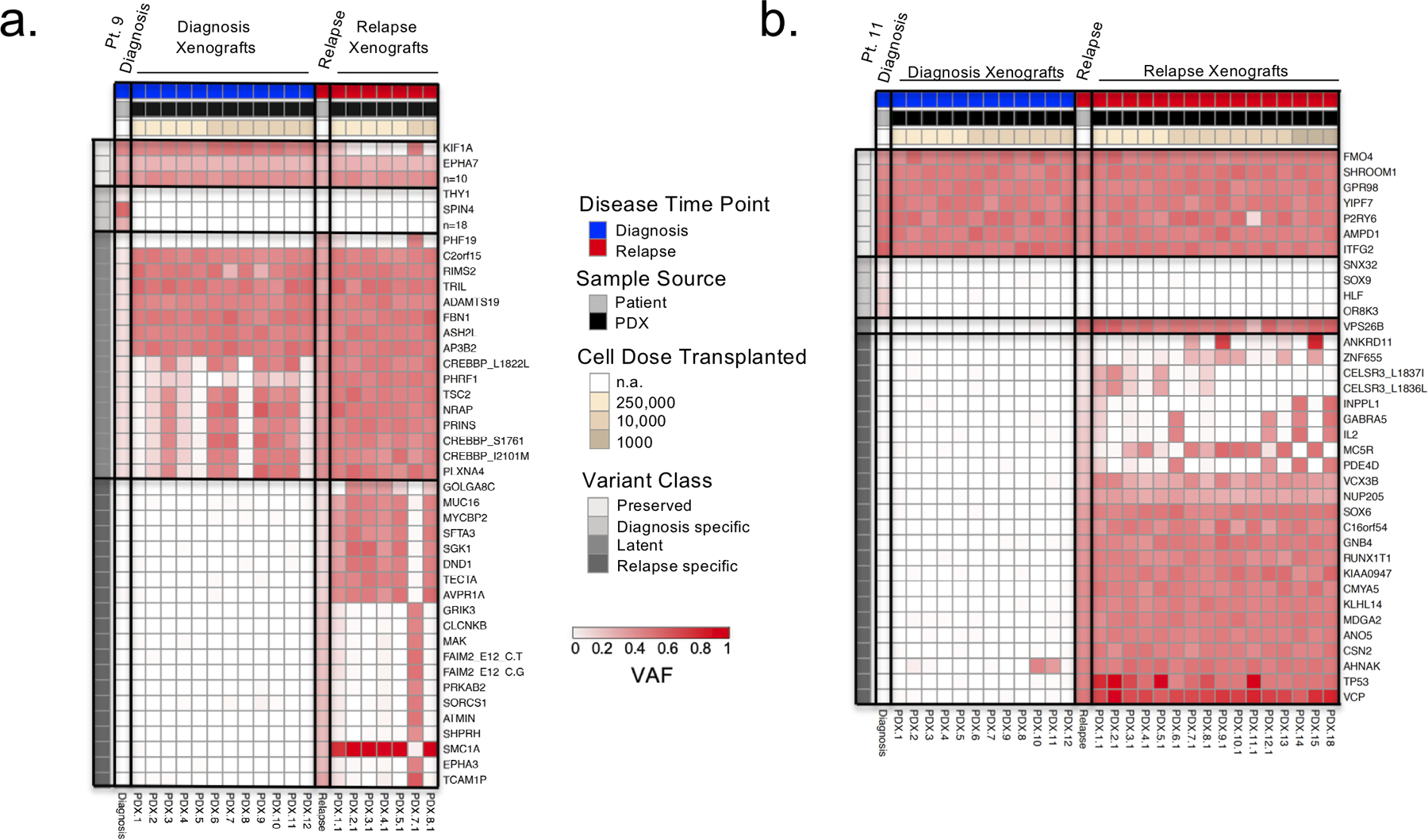
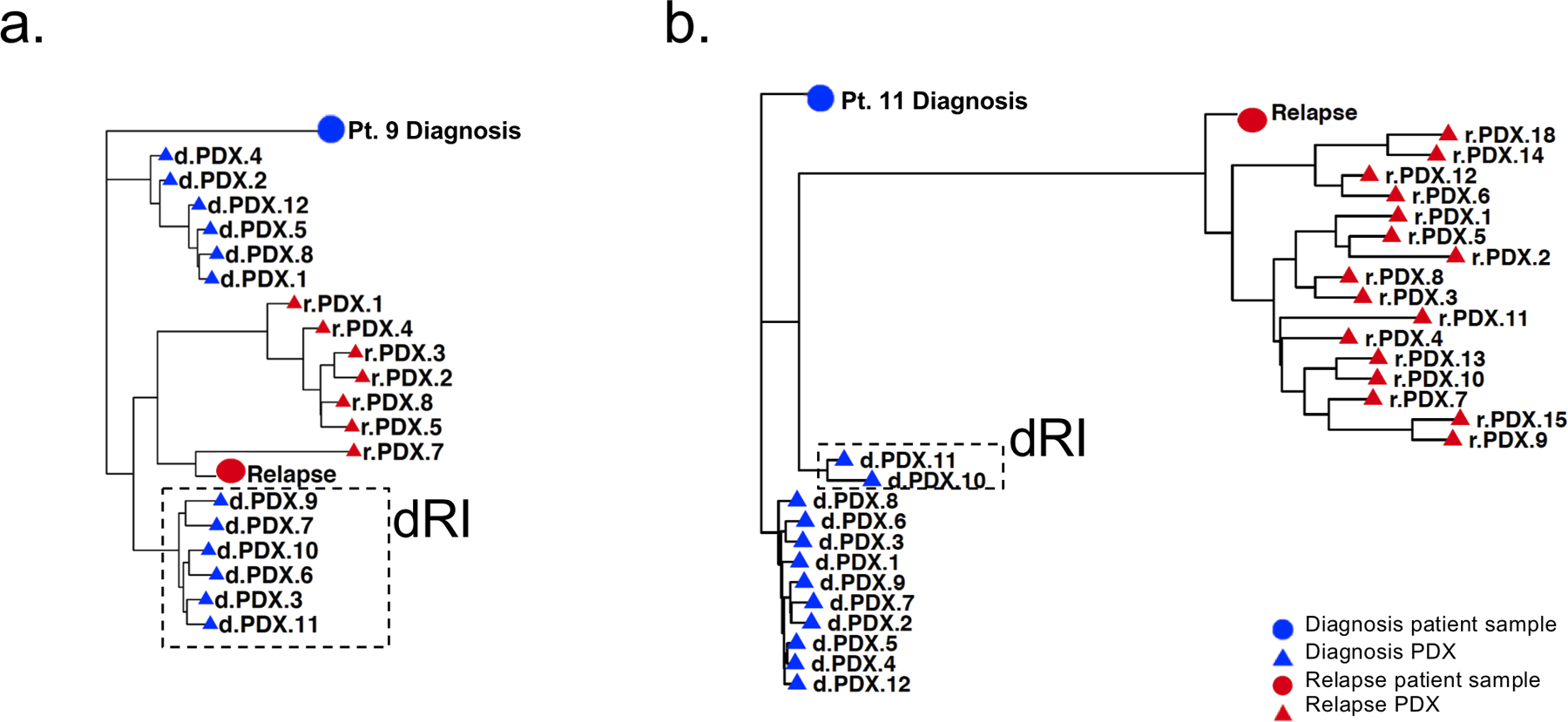
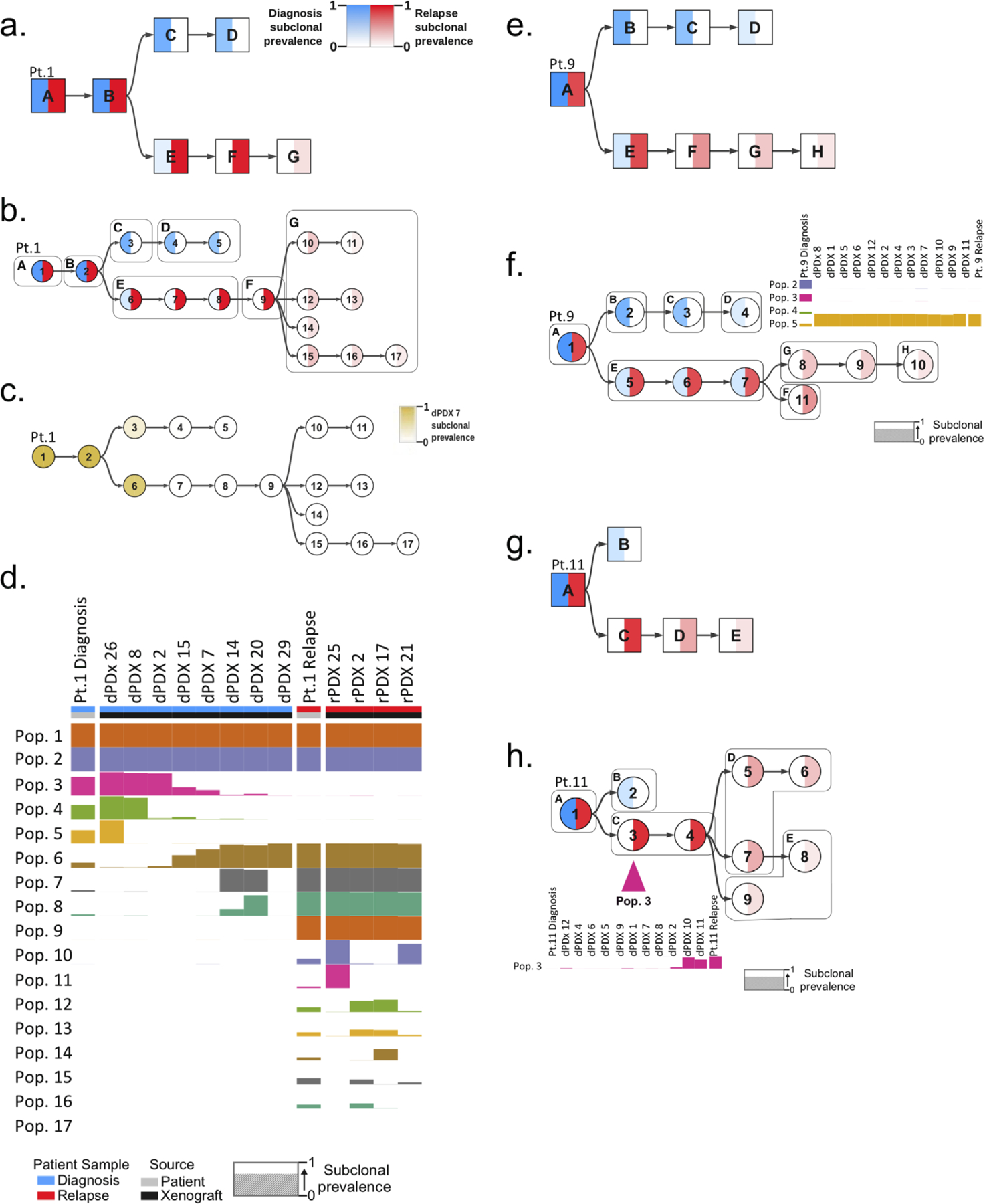
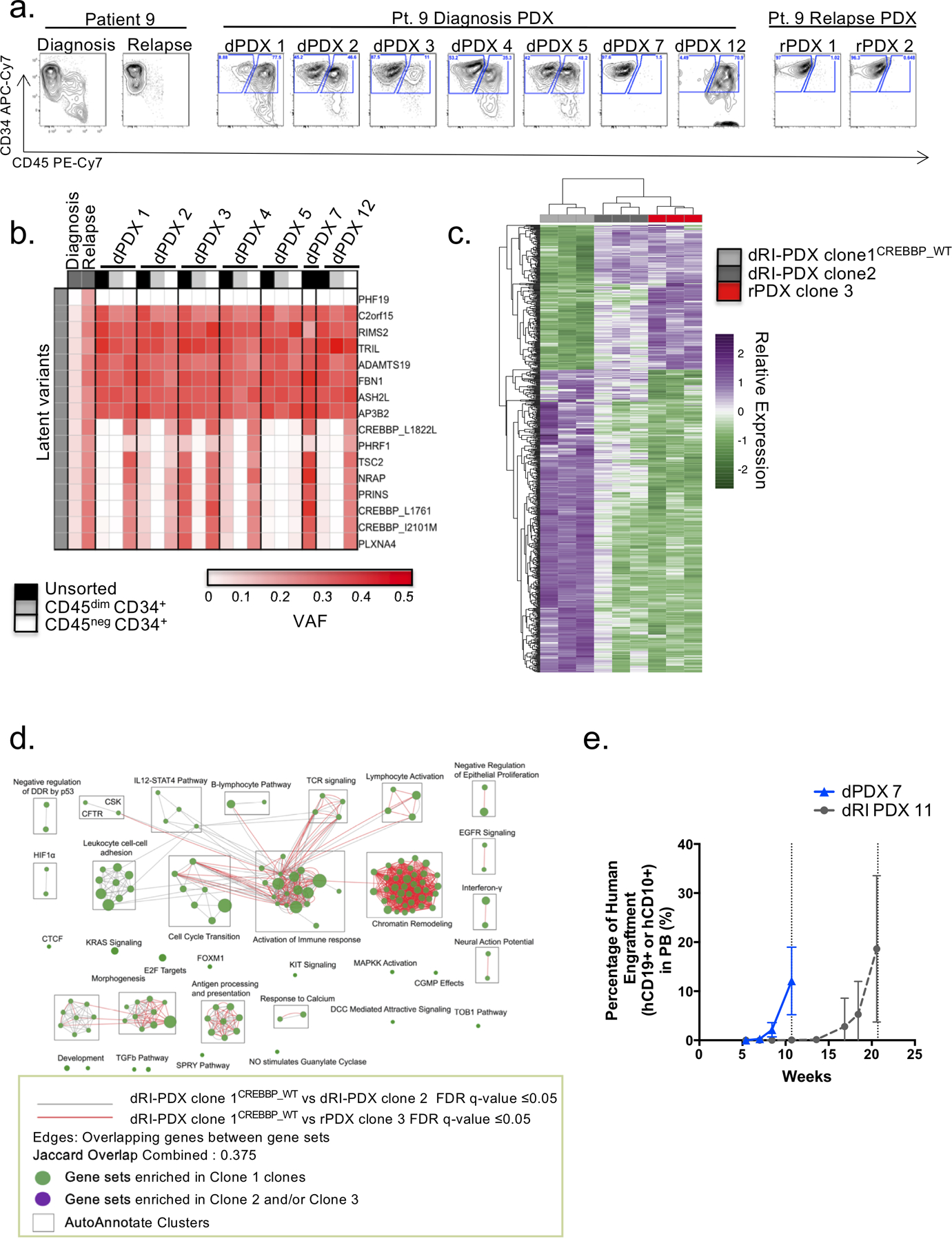
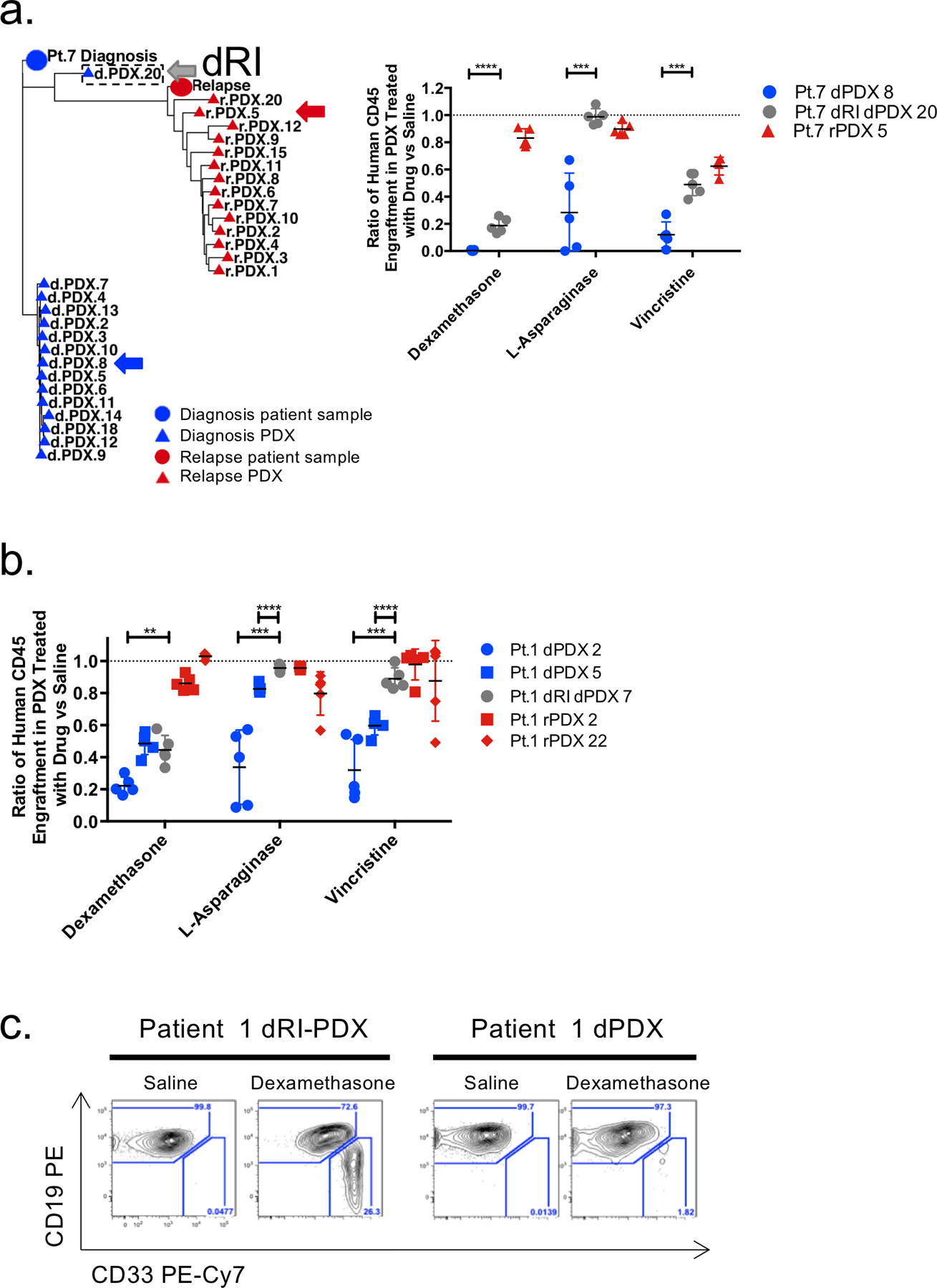
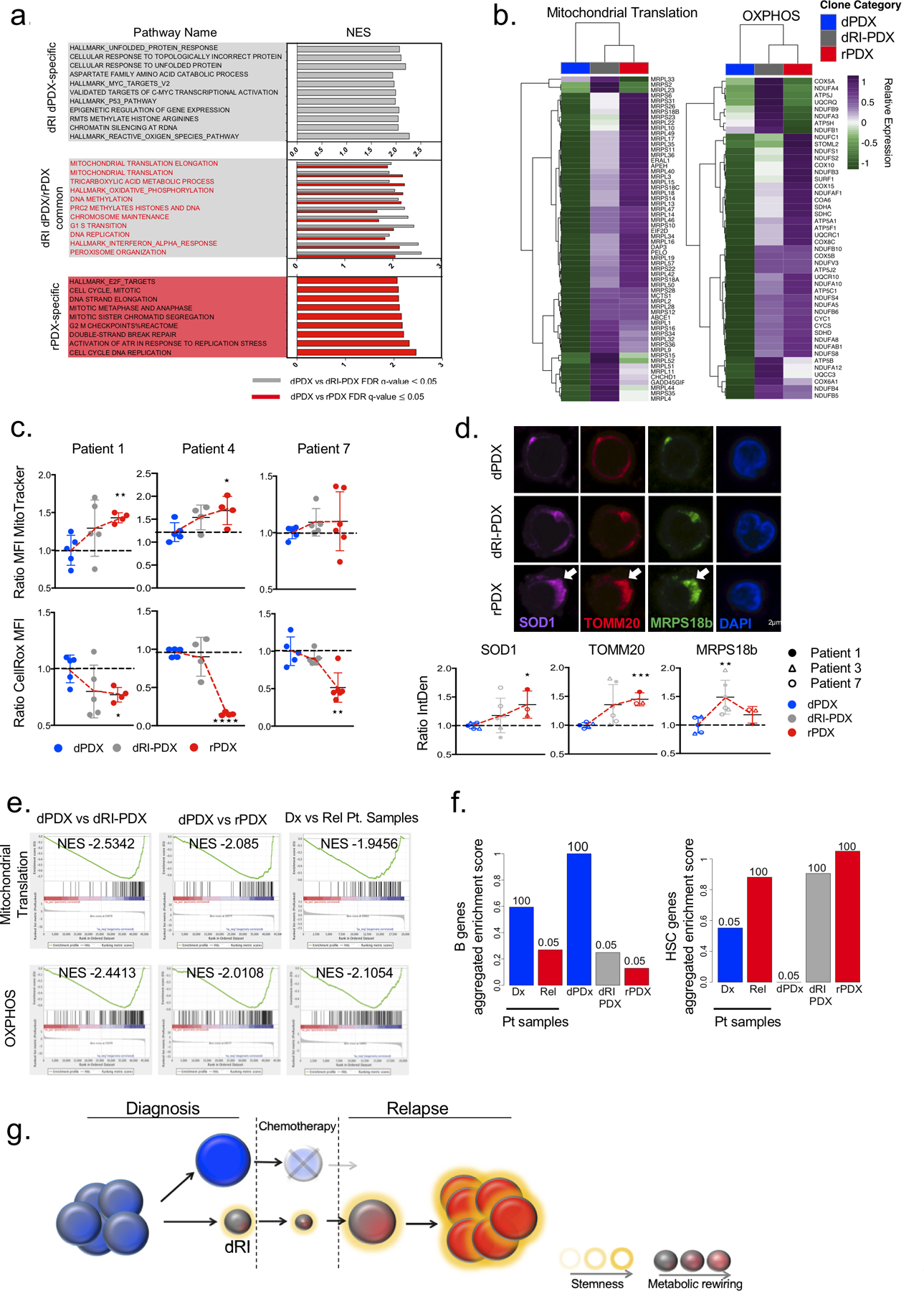
Disease recurrence causes significant mortality in B-progenitor acute lymphoblastic leukemia (B-ALL). Genomic analysis of matched diagnosis and relapse samples shows relapse often arising from minor diagnosis subclones. However, why therapy eradicates some subclones while others survive and progress to relapse remains obscure. Elucidation of mechanisms underlying these differing fates requires functional analysis of isolated subclones. Here, large-scale limiting dilution xenografting of diagnosis and relapse samples, combined with targeted sequencing, identified and isolated minor diagnosis subclones that initiate an evolutionary trajectory toward relapse [termed diagnosis Relapse Initiating clones (dRI)]. Compared with other diagnosis subclones, dRIs were drug-tolerant with distinct engraftment and metabolic properties. Transcriptionally, dRIs displayed enrichment for chromatin remodeling, mitochondrial metabolism, proteostasis programs, and an increase in stemness pathways. The isolation and characterization of dRI subclones reveals new avenues for eradicating dRI cells by targeting their distinct metabolic and transcriptional pathways before further evolution renders them fully therapy-resistant. SIGNIFICANCE: Isolation and characterization of subclones from diagnosis samples of patients with B-ALL who relapsed showed that relapse-fated subclones had increased drug tolerance and distinct metabolic and survival transcriptional programs compared with other diagnosis subclones. This study provides strategies to identify and target clinically relevant subclones before further evolution toward relapse.
©2020 American Association for Cancer Research.
Conflict of interest statement
Conflict of Interest:
J.E.D. Celgene: research funding; Trillium Therapeutics: Advisory Board funding; Abbvie: research funding
C.G.M. Illumina: Advisory board, honorarium, sponsored travel; Pfizer: research
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
