

MET Alterations Are a Recurring and Actionable Resistance Mechanism in ALK-Positive Lung Cancer
- PMID: 32086345
- PMCID: PMC7269872
- DOI: 10.1158/1078-0432.CCR-19-3906
MET Alterations Are a Recurring and Actionable Resistance Mechanism in ALK-Positive Lung Cancer
Abstract
Purpose: Most ALK-positive lung cancers will develop ALK-independent resistance after treatment with next-generation ALK inhibitors. MET amplification has been described in patients progressing on ALK inhibitors, but frequency of this event has not been comprehensively assessed.
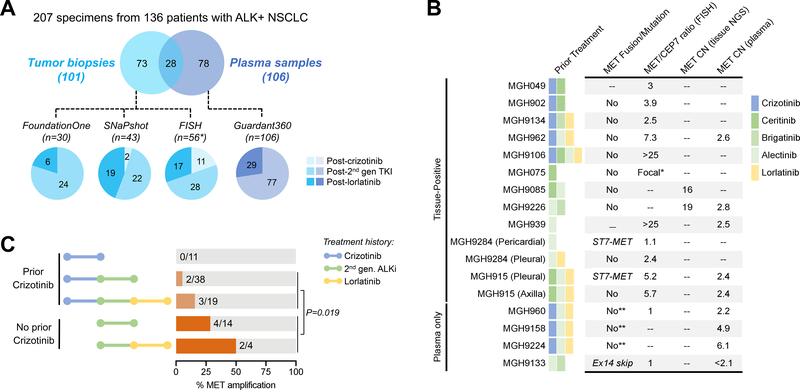
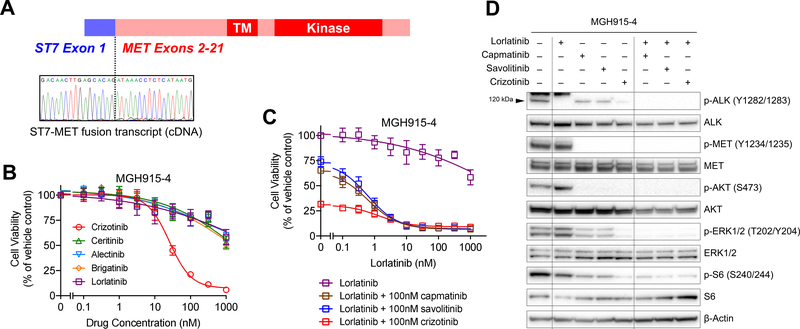
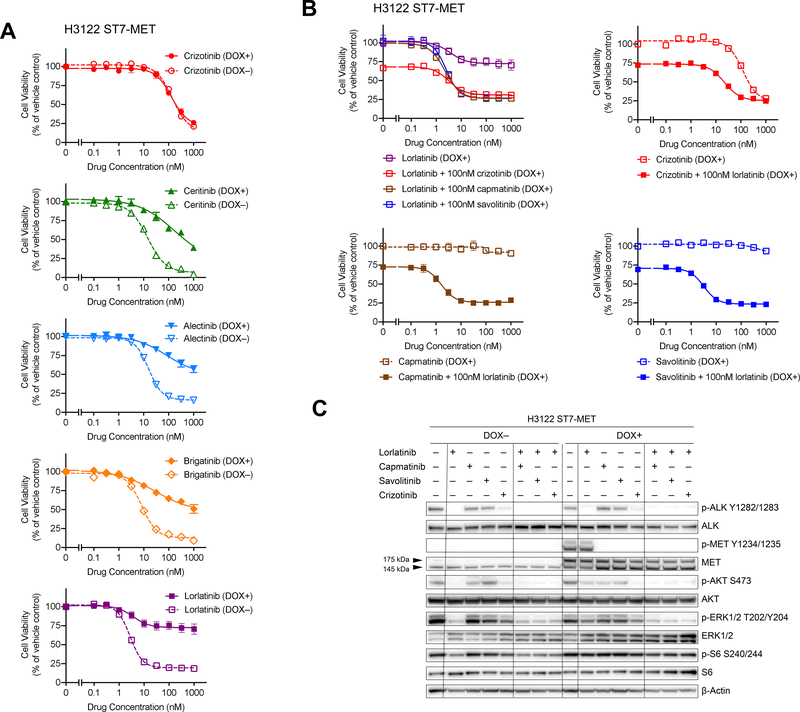
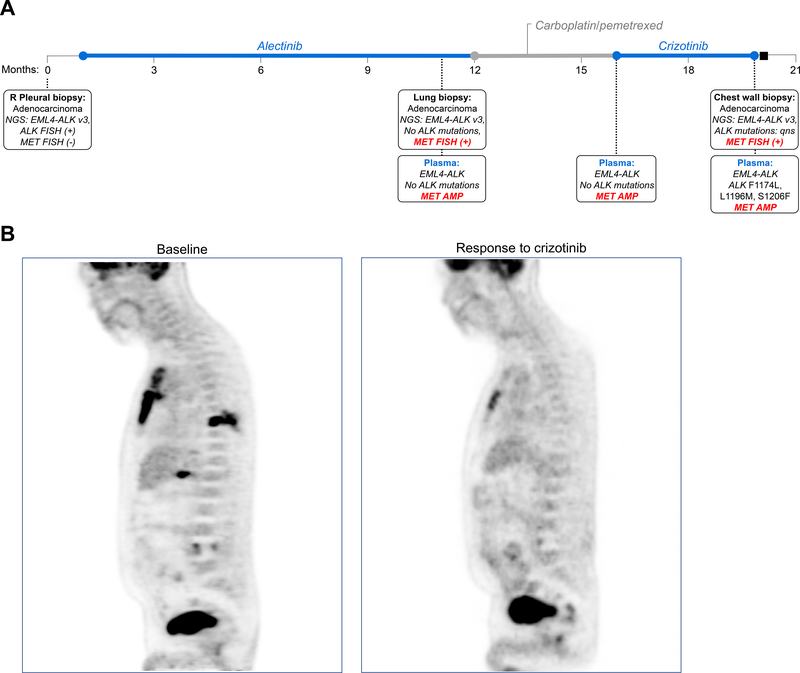
Experimental design: We performed FISH and/or next-generation sequencing on 207 posttreatment tissue (n = 101) or plasma (n = 106) specimens from patients with ALK-positive lung cancer to detect MET genetic alterations. We evaluated ALK inhibitor sensitivity in cell lines with MET alterations and assessed antitumor activity of ALK/MET blockade in ALK-positive cell lines and 2 patients with MET-driven resistance.
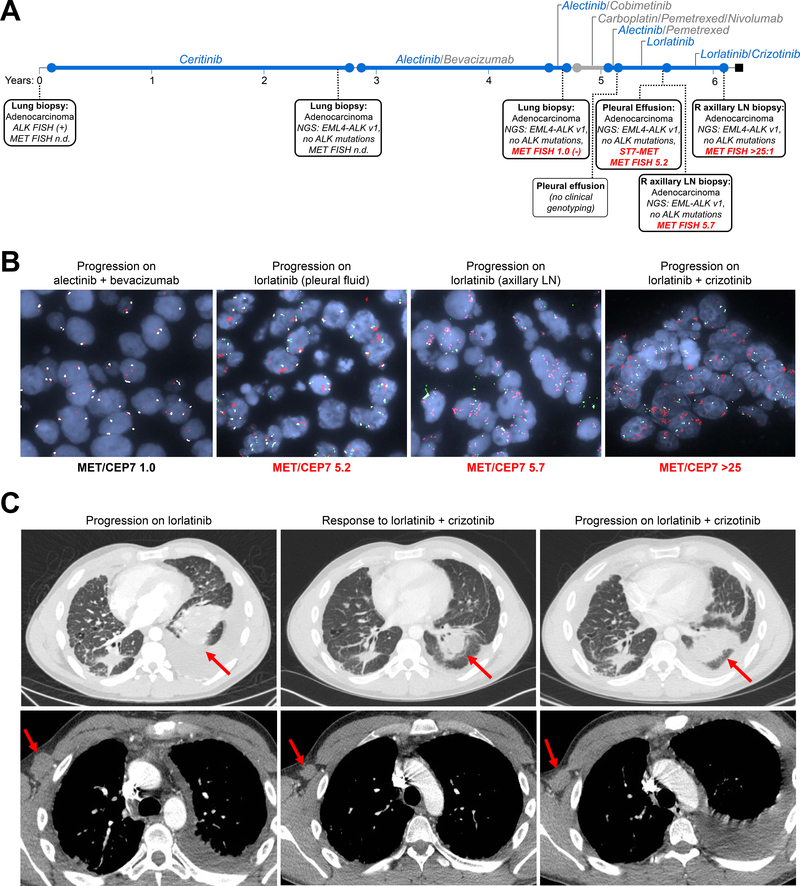
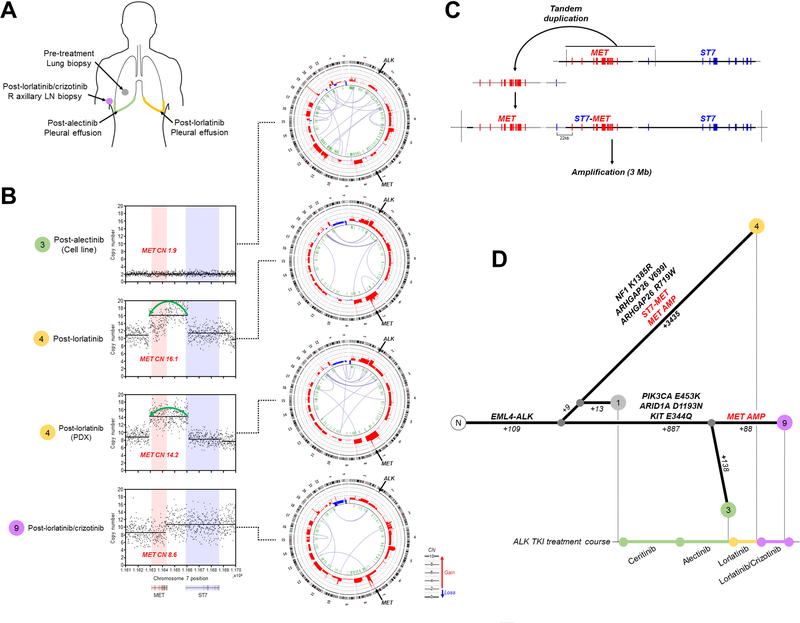
Results: MET amplification was detected in 15% of tumor biopsies from patients relapsing on next-generation ALK inhibitors, including 12% and 22% of biopsies from patients progressing on second-generation inhibitors or lorlatinib, respectively. Patients treated with a second-generation ALK inhibitor in the first-line setting were more likely to develop MET amplification than those who had received next-generation ALK inhibitors after crizotinib (P = 0.019). Two tumor specimens harbored an identical ST7-MET rearrangement, one of which had concurrent MET amplification. Expressing ST7-MET in the sensitive H3122 ALK-positive cell line induced resistance to ALK inhibitors that was reversed with dual ALK/MET inhibition. MET inhibition resensitized a patient-derived cell line harboring both ST7-MET and MET amplification to ALK inhibitors. Two patients with ALK-positive lung cancer and acquired MET alterations achieved rapid responses to ALK/MET combination therapy.
Conclusions: Treatment with next-generation ALK inhibitors, particularly in the first-line setting, may lead to MET-driven resistance. Patients with acquired MET alterations may derive clinical benefit from therapies that target both ALK and MET.
©2020 American Association for Cancer Research.
Figures
References
-
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. - PubMed
-
- Camidge DR, Kim HR, Ahn MJ, et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2018;379(21):2027–2039. - PubMed
-
- Peters S, Camidge DR, Shaw AT, et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N Engl J Med. 2017;377(9):829–838. - PubMed
-
- Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. - PubMed
-
- Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet. 2017;389(10072):917–929. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
