

cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons
- PMID: 32087251
- PMCID: PMC7302925
- DOI: 10.1016/j.expneurol.2020.113252
cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons
Abstract
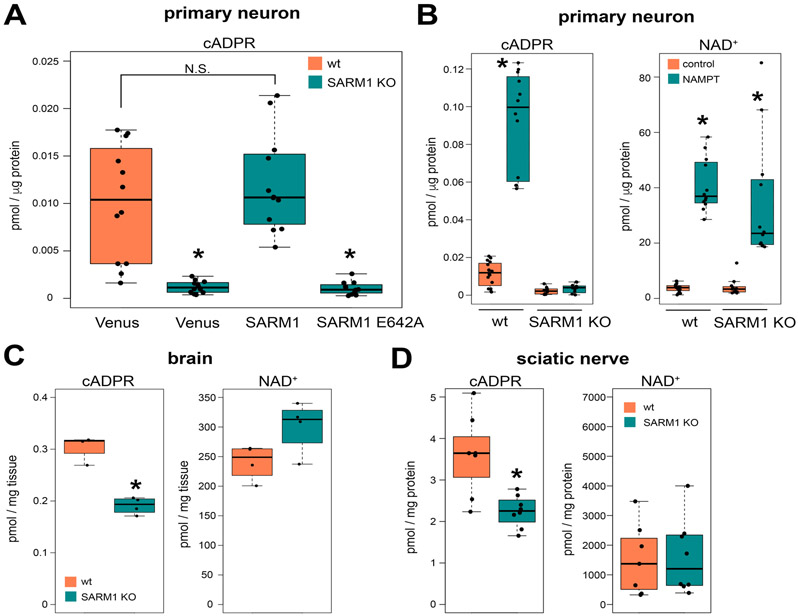
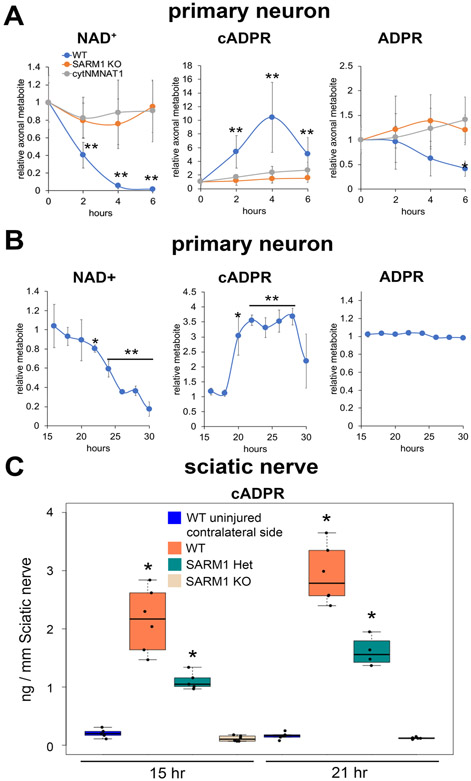
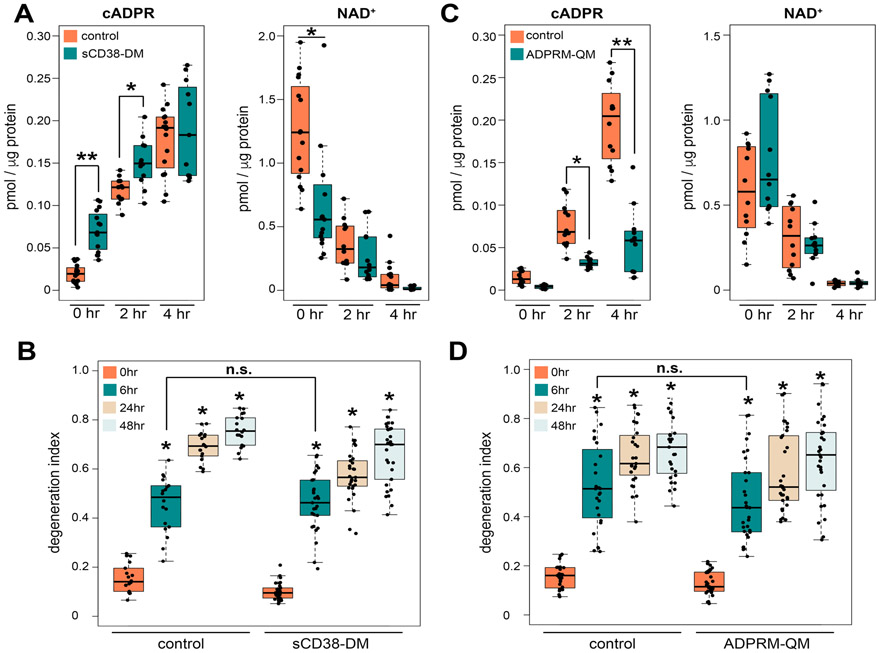
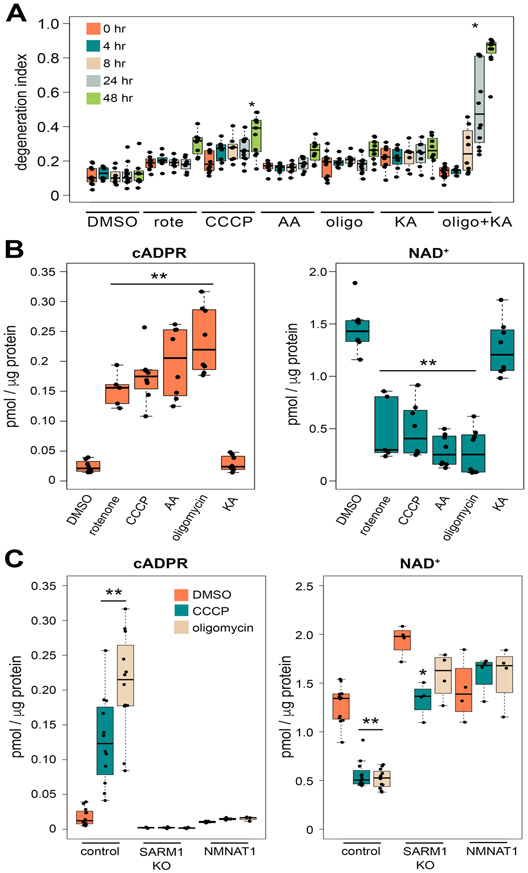
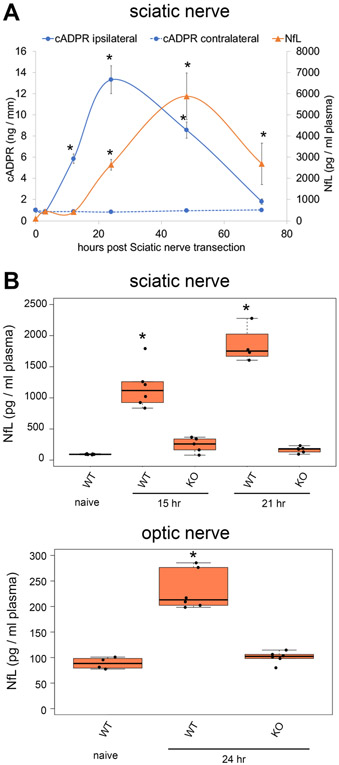
SARM1 is the central executioner of pathological axon degeneration, promoting axonal demise in response to axotomy, traumatic brain injury, and neurotoxic chemotherapeutics that induce peripheral neuropathy. SARM1 is an injury-activated NAD+ cleavage enzyme, and this NADase activity is required for the pro-degenerative function of SARM1. At present, SARM1 function is assayed by either analysis of axonal loss, which is far downstream of SARM1 enzymatic activity, or via NAD+ levels, which are regulated by many competing pathways. Here we explored the utility of measuring cADPR, a product of SARM1-dependent cleavage of NAD+, as an in cell and in vivo biomarker of SARM1 enzymatic activity. We find that SARM1 is a major producer of cADPR in cultured dorsal root ganglion (DRG) neurons, sciatic nerve, and brain, demonstrating that SARM1 has basal activity in the absence of injury. Following injury, there is a dramatic SARM1-dependent increase in the levels of axonal cADPR that precedes morphological axon degeneration. In vivo, there is also a rapid and large injury-stimulated increase in cADPR in sciatic and optic nerves. The increase in cADPR after injury is proportional to SARM1 gene dosage, suggesting that SARM1 activity is the prime regulator of cADPR levels. The role of cADPR as an important calcium mobilizing agent prompted exploration of its functional contribution to axon degeneration. We used multiple bacterial and mammalian engineered enzymes to manipulate cADPR levels in neurons but found no changes in the time course of axonal degeneration, suggesting that cADPR is unlikely to be an important contributor to the degenerative mechanism. Using cADPR as a SARM1 biomarker, we find that SARM1 can be partially activated by a diverse array of mitochondrial toxins administered at doses that do not induce axon degeneration. Hence, the subcritical activation of SARM1 induced by mitochondrial dysfunction may contribute to the axonal vulnerability common to many neurodegenerative diseases. Finally, we assay levels of both nerve cADPR and plasma neurofilament light chain (NfL) following nerve injury in vivo, and demonstrate that both biomarkers are excellent readouts of SARM1 activity, with cADPR reporting the early molecular changes in the nerve and NfL reporting subsequent axonal breakdown. The identification and characterization of cADPR as a SARM1 biomarker will help identify neurodegenerative diseases in which SARM1 contributes to axonal loss and expedite target validation studies of SARM1-directed therapeutics.
Copyright © 2020. Published by Elsevier Inc.
Conflict of interest statement
Declaration of Competing Interest A.D and J.M. co-founders of and shareholders in Disarm Therapeutics. Y.S. is a consultant to Disarm Therapeutics. R.K., R.O.H., T.M.E., T.B. and R.D. are employees and shareholders of Disarm Therapeutics. The authors have no additional competing financial interests.
Figures
Similar articles
-
Gap junction intercellular communications regulates activation of SARM1 and protects against axonal degeneration.Cell Death Dis. 2025 Jan 14;16(1):13. doi: 10.1038/s41419-025-07342-4. Cell Death Dis. 2025. PMID: 39809779 Free PMC article.
-
The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration.Neuron. 2017 Mar 22;93(6):1334-1343.e5. doi: 10.1016/j.neuron.2017.02.022. Neuron. 2017. PMID: 28334607 Free PMC article.
-
SARM1 is required in human derived sensory neurons for injury-induced and neurotoxic axon degeneration.Exp Neurol. 2021 May;339:113636. doi: 10.1016/j.expneurol.2021.113636. Epub 2021 Feb 4. Exp Neurol. 2021. PMID: 33548217 Free PMC article.
-
SARM1-Dependent Axon Degeneration: Nucleotide Signaling, Neurodegenerative Disorders, Toxicity, and Therapeutic Opportunities.Neuroscientist. 2024 Aug;30(4):473-492. doi: 10.1177/10738584231162508. Epub 2023 Mar 31. Neuroscientist. 2024. PMID: 37002660 Free PMC article. Review.
-
SARM1: The Checkpoint of Axonal Degeneration in the Nervous System Disorders.Mol Neurobiol. 2025 Jul;62(7):9240-9257. doi: 10.1007/s12035-025-04835-3. Epub 2025 Mar 17. Mol Neurobiol. 2025. PMID: 40097763 Review.
Cited by
-
SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration.J Cell Biol. 2020 Aug 3;219(8):e201912047. doi: 10.1083/jcb.201912047. J Cell Biol. 2020. PMID: 32609299 Free PMC article.
-
Loss of Stathmin-2, a hallmark of TDP-43-associated ALS, causes motor neuropathy.Cell Rep. 2022 Jun 28;39(13):111001. doi: 10.1016/j.celrep.2022.111001. Cell Rep. 2022. PMID: 35767949 Free PMC article.
-
SARM1 is an essential component of neuronal Parthanatos.bioRxiv [Preprint]. 2025 May 15:2025.05.14.654090. doi: 10.1101/2025.05.14.654090. bioRxiv. 2025. PMID: 40463025 Free PMC article. Preprint.
-
Suppressing phagocyte activation by overexpressing the phosphatidylserine lipase ABHD12 preserves sarmopathic nerves.iScience. 2025 May 9;28(6):112626. doi: 10.1016/j.isci.2025.112626. eCollection 2025 Jun 20. iScience. 2025. PMID: 40496808 Free PMC article.
-
Multiple domain interfaces mediate SARM1 autoinhibition.Proc Natl Acad Sci U S A. 2021 Jan 26;118(4):e2023151118. doi: 10.1073/pnas.2023151118. Proc Natl Acad Sci U S A. 2021. PMID: 33468661 Free PMC article.
References
-
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004; 305: 1010–1013. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
