

Succinic Semialdehyde Dehydrogenase Deficiency: An Update
- PMID: 32093054
- PMCID: PMC7072817
- DOI: 10.3390/cells9020477
Succinic Semialdehyde Dehydrogenase Deficiency: An Update
Abstract
Succinic semialdehyde dehydrogenase deficiency (SSADH-D) is a genetic disorder that results from the aberrant metabolism of the neurotransmitter γ-amino butyric acid (GABA). The disease is caused by impaired activity of the mitochondrial enzyme succinic semialdehyde dehydrogenase. SSADH-D manifests as varying degrees of mental retardation, autism, ataxia, and epileptic seizures, but the clinical picture is highly heterogeneous. So far, there is no approved curative therapy for this disease. In this review, we briefly summarize the molecular genetics of SSADH-D, the past and ongoing clinical trials, and the emerging features of the molecular pathogenesis, including redox imbalance and mitochondrial dysfunction. The main aim of this review is to discuss the potential of further therapy approaches that have so far not been tested in SSADH-D, such as pharmacological chaperones, read-through drugs, and gene therapy. Special attention will also be paid to elucidating the role of patient advocacy organizations in facilitating research and in the communication between researchers and patients.
Keywords: autophagy; clinical trials; enzyme replacement therapy; gamma-amino butyric acid; organic acidurias; pharmacological chaperones; succinic semialdehyde dehydrogenase deficiency.
Conflict of interest statement
The authors T.O., H.B. and R.T. declare to have received funding from SSADH-Defizit e.V, a patient advocacy organization. The funders had no role in the decision to publish the article.
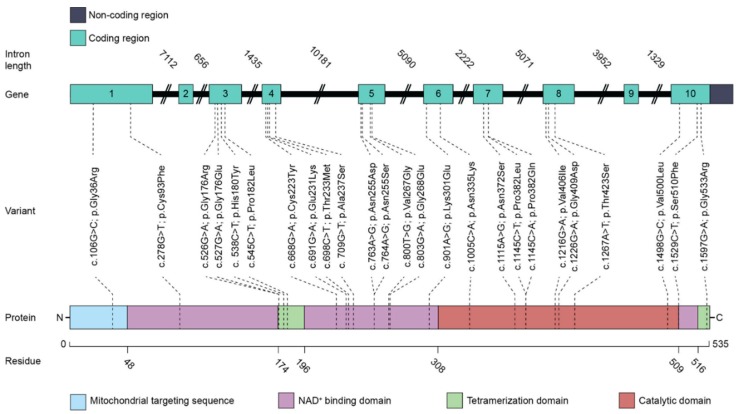
Figures


References
-
- Gibson K.M., Jakobs C. Disorders of beta- and alpha-amino acids in free and peptide-linked forms. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., Childs B., Kinzler K.W., Vogelstein B., editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. McGraw-Hill; New York, NY, USA: 2001. pp. 2079–2105.
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
