Contribution of common and rare variants to bipolar disorder susceptibility in extended pedigrees from population isolates
- PMID: 32094344
- PMCID: PMC7039961
- DOI: 10.1038/s41398-020-0758-1
Contribution of common and rare variants to bipolar disorder susceptibility in extended pedigrees from population isolates
Abstract
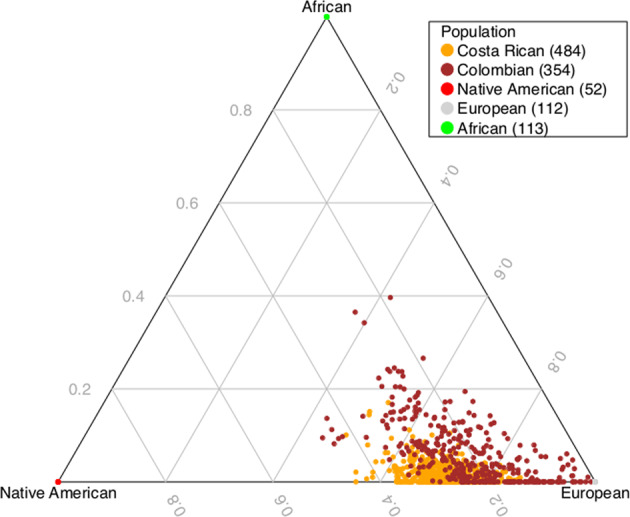
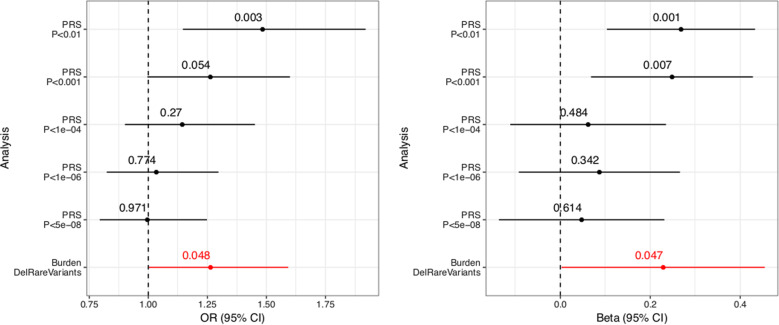
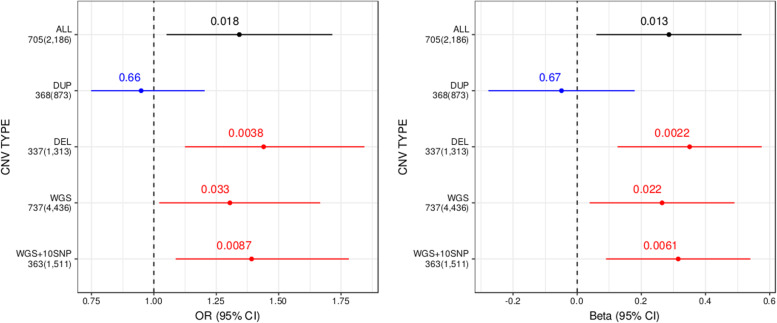
Current evidence from case/control studies indicates that genetic risk for psychiatric disorders derives primarily from numerous common variants, each with a small phenotypic impact. The literature describing apparent segregation of bipolar disorder (BP) in numerous multigenerational pedigrees suggests that, in such families, large-effect inherited variants might play a greater role. To identify roles of rare and common variants on BP, we conducted genetic analyses in 26 Colombia and Costa Rica pedigrees ascertained for bipolar disorder 1 (BP1), the most severe and heritable form of BP. In these pedigrees, we performed microarray SNP genotyping of 838 individuals and high-coverage whole-genome sequencing of 449 individuals. We compared polygenic risk scores (PRS), estimated using the latest BP1 genome-wide association study (GWAS) summary statistics, between BP1 individuals and related controls. We also evaluated whether BP1 individuals had a higher burden of rare deleterious single-nucleotide variants (SNVs) and rare copy number variants (CNVs) in a set of genes related to BP1. We found that compared with unaffected relatives, BP1 individuals had higher PRS estimated from BP1 GWAS statistics (P = 0.001 ~ 0.007) and displayed modest increase in burdens of rare deleterious SNVs (P = 0.047) and rare CNVs (P = 0.002 ~ 0.033) in genes related to BP1. We did not observe rare variants segregating in the pedigrees. These results suggest that small-to-moderate effect rare and common variants are more likely to contribute to BP1 risk in these extended pedigrees than a few large-effect rare variants.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures