

A drug discovery platform to identify compounds that inhibit EGFR triple mutants
- PMID: 32094923
- PMCID: PMC8123931
- DOI: 10.1038/s41589-020-0484-2
A drug discovery platform to identify compounds that inhibit EGFR triple mutants
Abstract
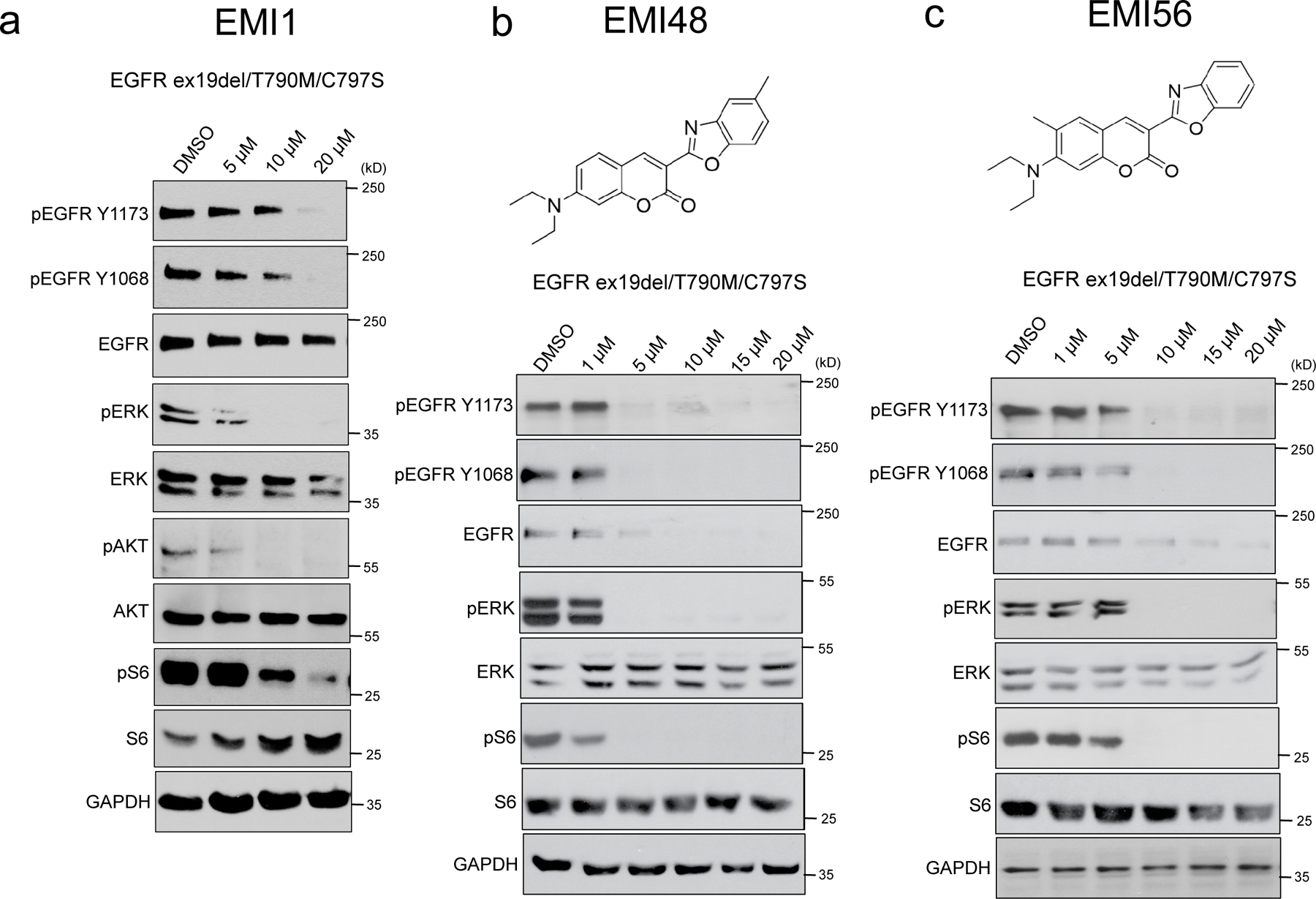
Receptor tyrosine kinases (RTKs) are transmembrane receptors of great clinical interest due to their role in disease. Historically, therapeutics targeting RTKs have been identified using in vitro kinase assays. Due to frequent development of drug resistance, however, there is a need to identify more diverse compounds that inhibit mutated but not wild-type RTKs. Here, we describe MaMTH-DS (mammalian membrane two-hybrid drug screening), a live-cell platform for high-throughput identification of small molecules targeting functional protein-protein interactions of RTKs. We applied MaMTH-DS to an oncogenic epidermal growth factor receptor (EGFR) mutant resistant to the latest generation of clinically approved tyrosine kinase inhibitors (TKIs). We identified four mutant-specific compounds, including two that would not have been detected by conventional in vitro kinase assays. One of these targets mutant EGFR via a new mechanism of action, distinct from classical TKI inhibition. Our results demonstrate how MaMTH-DS is a powerful complement to traditional drug screening approaches.
Conflict of interest statement
COMPETING FINANCIAL INTERESTS
I.S., P.S. and J.S. (in conjunction with the University of Toronto) are listed as inventors on a patent (publication number 20190091205) for the use of EMI1 (and structurally related analogues), midostaurin, gilteritinib and AZD7762 (and structurally related analogues) in the treatment of mutant EGFR-mediated non-small-cell lung cancer.
Figures
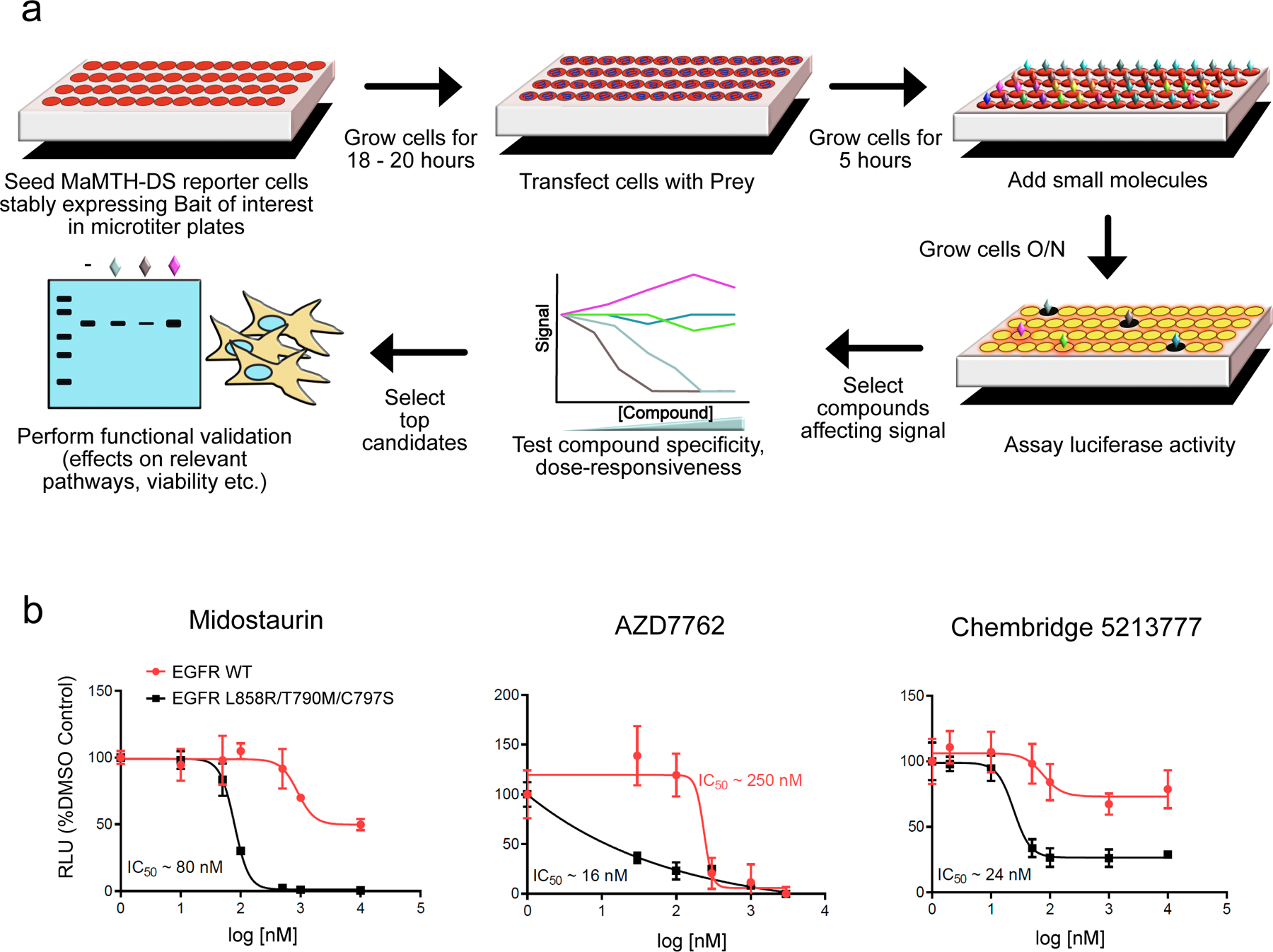
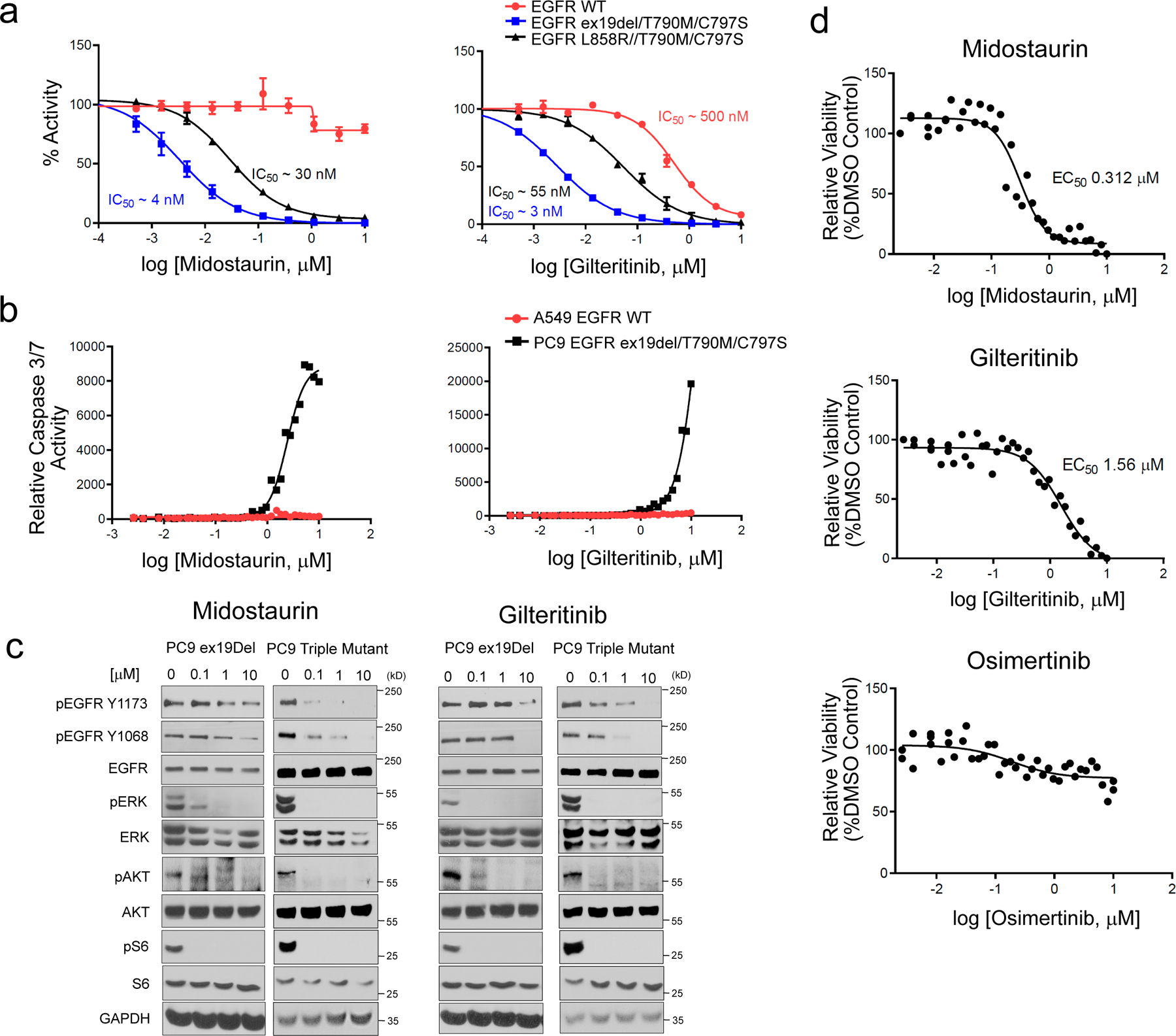
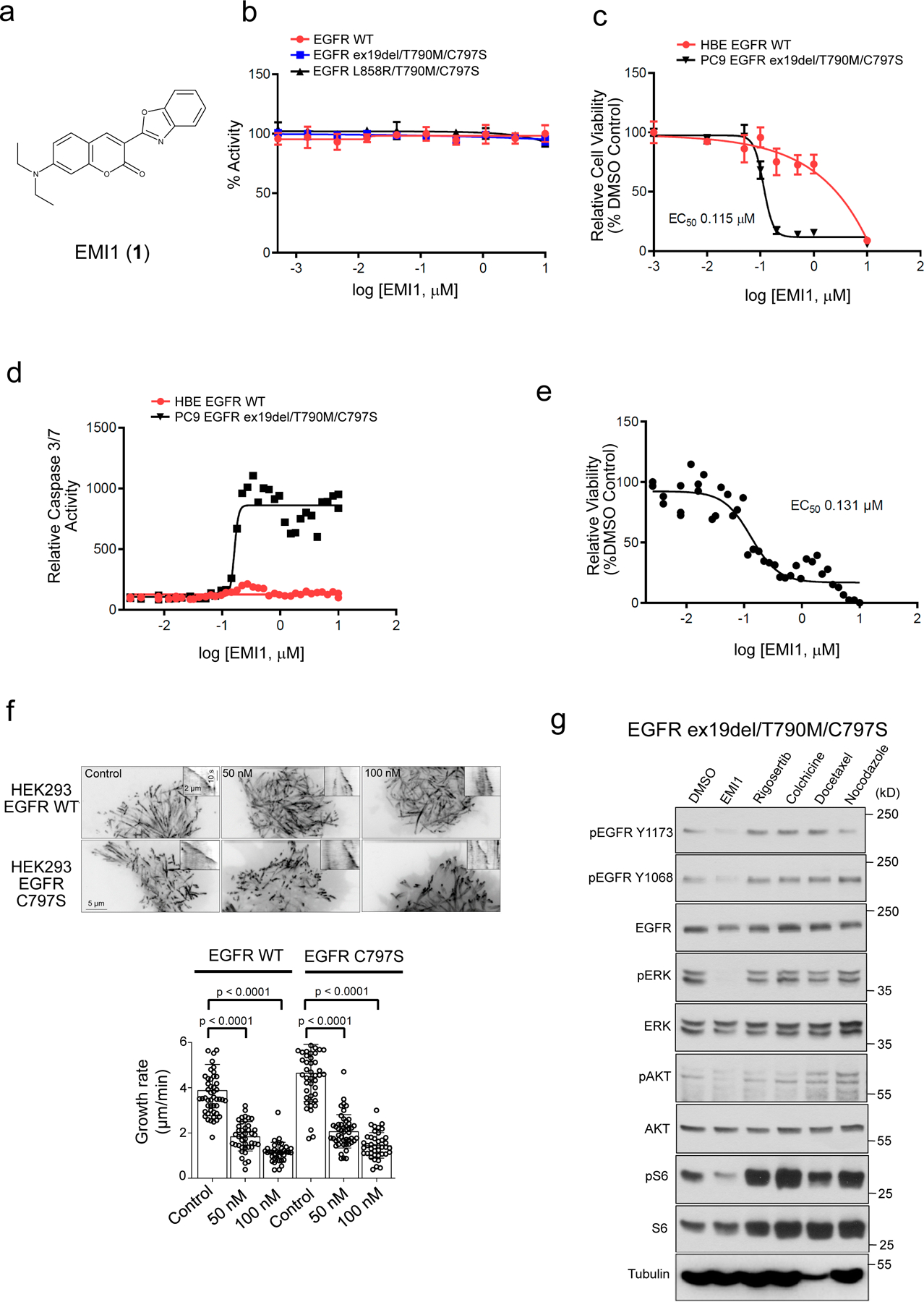
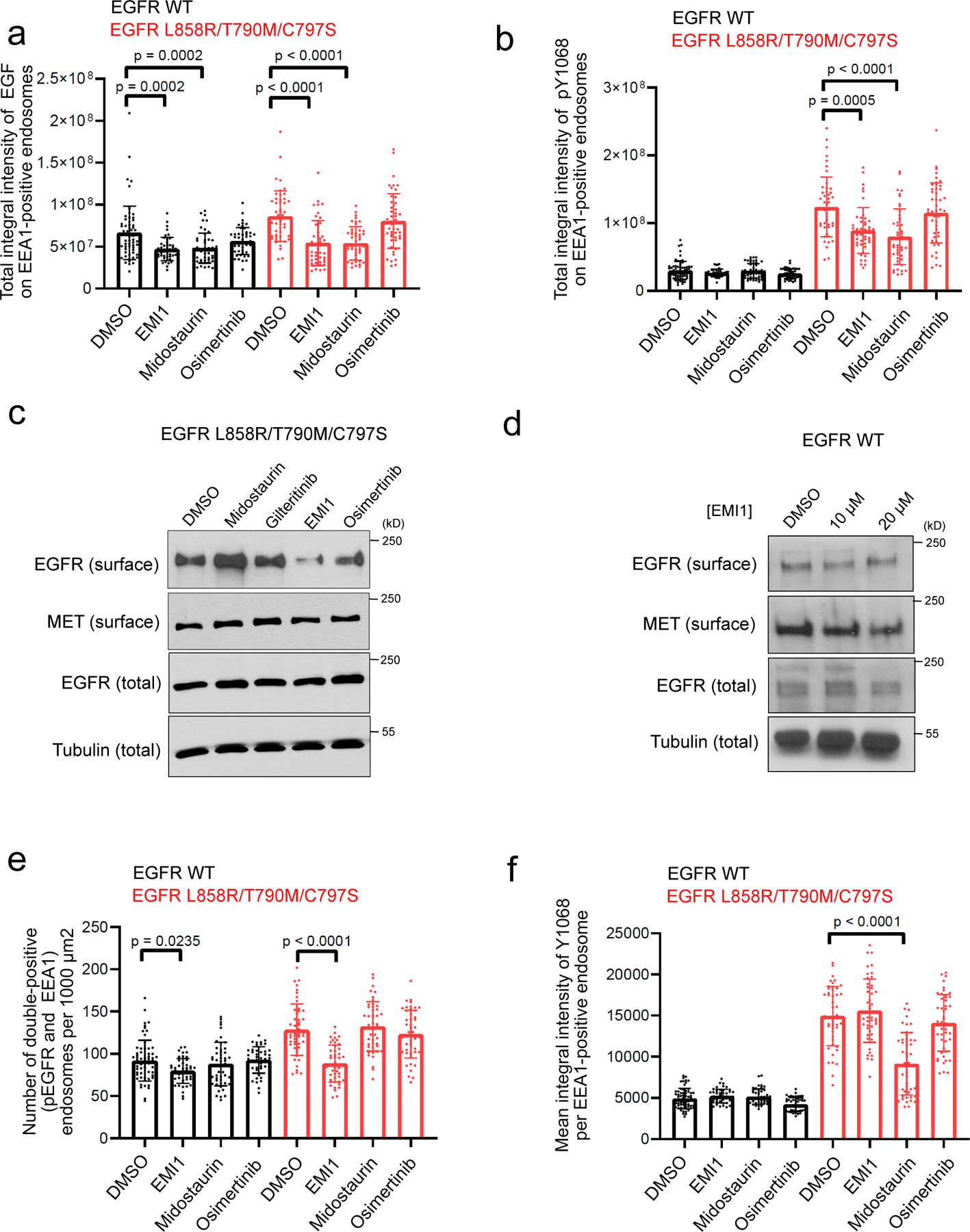
Comment in
-
New Drug-Discovery Assay Identifies Novel Mutant-EGFR Inhibitors.Cancer Discov. 2020 May;10(5):635. doi: 10.1158/2159-8290.CD-RW2020-035. Epub 2020 Mar 6. Cancer Discov. 2020. PMID: 32144093
References
-
- Jia Y, Quinn CM, Kwak S & Talanian RV Current in vitro kinase assay technologies: the quest for a universal format. Curr. Drug Discov. Technol 5, 59–69 (2008). - PubMed
-
- Petschnigg J et al. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat. Methods 11, 585–92 (2014). - PubMed
METHODS-ONLY REFERENCES
-
- Collinet C et al. Systems survey of endocytosis by multiparametric image analysis. Nature 464, 243–9 (2010). - PubMed
-
- Rink J, Ghigo E, Kalaidzidis Y & Zerial M Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749 (2005). - PubMed
-
- Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). - PubMed
-
- R-Core-Team. R: A language and environment for statistical computing (2017).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
