

Insights into the pathogenesis of multiple system atrophy: focus on glial cytoplasmic inclusions
- PMID: 32095235
- PMCID: PMC7025408
- DOI: 10.1186/s40035-020-0185-5
Insights into the pathogenesis of multiple system atrophy: focus on glial cytoplasmic inclusions
Abstract
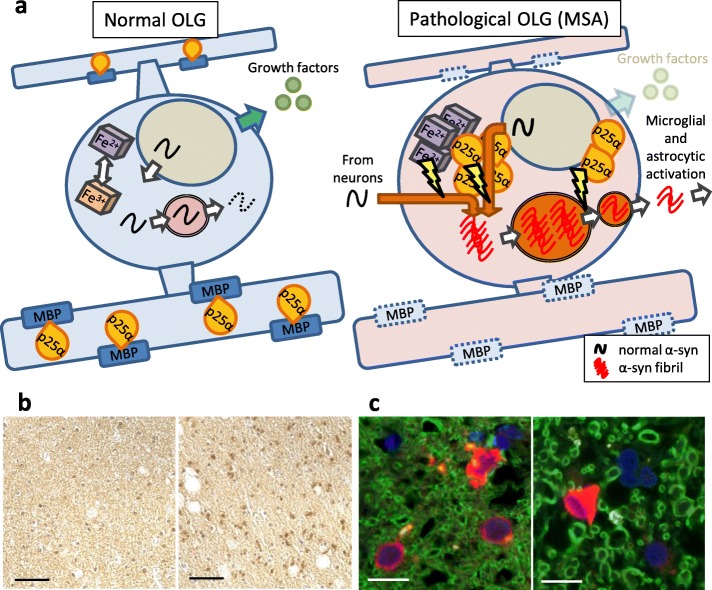
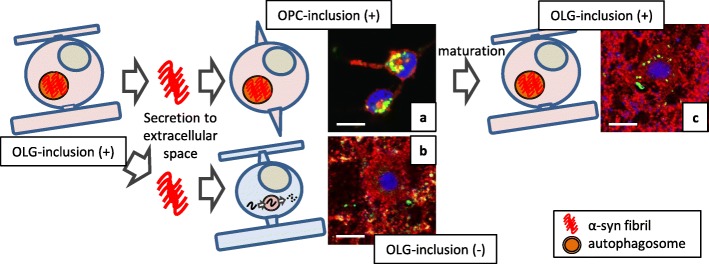
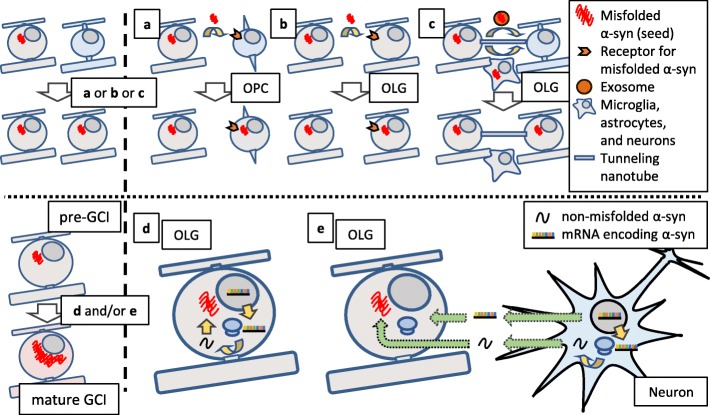
Multiple system atrophy (MSA) is a debilitating and fatal neurodegenerative disorder. The disease severity warrants urgent development of disease-modifying therapy, but the disease pathogenesis is still enigmatic. Neurodegeneration in MSA brains is preceded by the emergence of glial cytoplasmic inclusions (GCIs), which are insoluble α-synuclein accumulations within oligodendrocytes (OLGs). Thus, preventive strategies against GCI formation may suppress disease progression. However, although numerous studies have tried to elucidate the molecular pathogenesis of GCI formation, difficulty remains in understanding the pathological interaction between the two pivotal aspects of GCIs; α-synuclein and OLGs. The difficulty originates from several enigmas: 1) what triggers the initial generation and possible propagation of pathogenic α-synuclein species? 2) what contributes to OLG-specific accumulation of α-synuclein, which is abundantly expressed in neurons but not in OLGs? and 3) how are OLGs and other glial cells affected and contribute to neurodegeneration? The primary pathogenesis of GCIs may involve myelin dysfunction and dyshomeostasis of the oligodendroglial cellular environment such as autophagy and iron metabolism. We have previously reported that oligodendrocyte precursor cells are more prone to develop intracellular inclusions in the presence of extracellular fibrillary α-synuclein. This finding implies a possibility that the propagation of GCI pathology in MSA brains is mediated through the internalization of pathological α-synuclein into oligodendrocyte precursor cells. In this review, in order to discuss the pathogenesis of GCIs, we will focus on the composition of neuronal and oligodendroglial inclusions in synucleinopathies. Furthermore, we will introduce some hypotheses on how α-synuclein pathology spreads among OLGs in MSA brains, in the light of our data from the experiments with primary oligodendrocyte lineage cell culture. While various reports have focused on the mysterious source of α-synuclein in GCIs, insights into the mechanism which regulates the uptake of pathological α-synuclein into oligodendroglial cells may yield the development of the disease-modifying therapy for MSA. The interaction between glial cells and α-synuclein is also highlighted with previous studies of post-mortem human brains, cultured cells, and animal models, which provide comprehensive insight into GCIs and the MSA pathomechanisms.
Keywords: Astrocyte; Glial cytoplasmic inclusion; Microglia; Multiple system atrophy; Neurodegeneration; Oligodendrocyte; Oligodendrocyte precursor cell; Prion; α-Synuclein.
© The Author(s). 2020.
Conflict of interest statement
Competing interestsThe authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
