

Tau and Axonal Transport Misregulation in Tauopathies
- PMID: 32096030
- PMCID: PMC7099581
- DOI: 10.1007/978-981-32-9358-8_7
Tau and Axonal Transport Misregulation in Tauopathies
Abstract
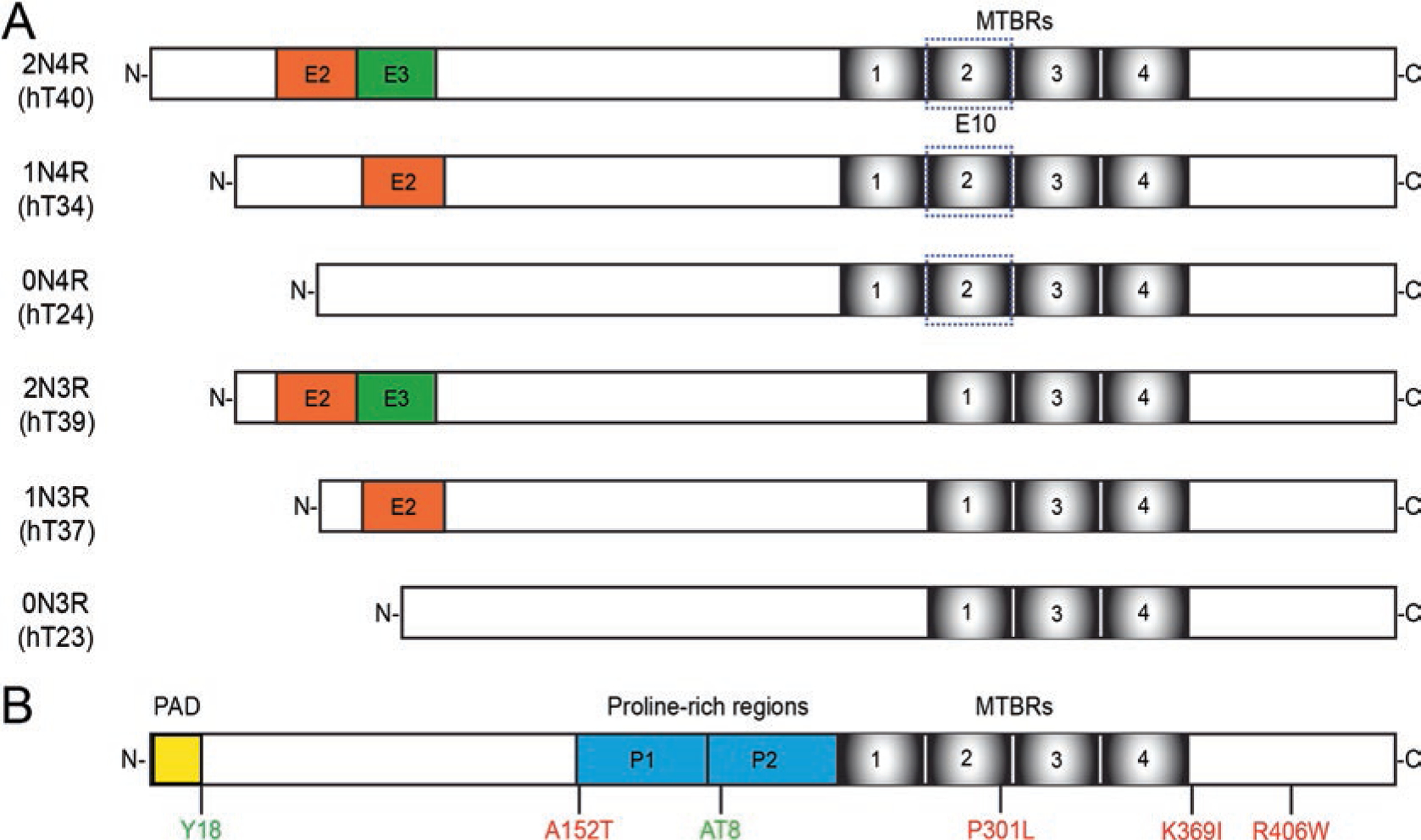
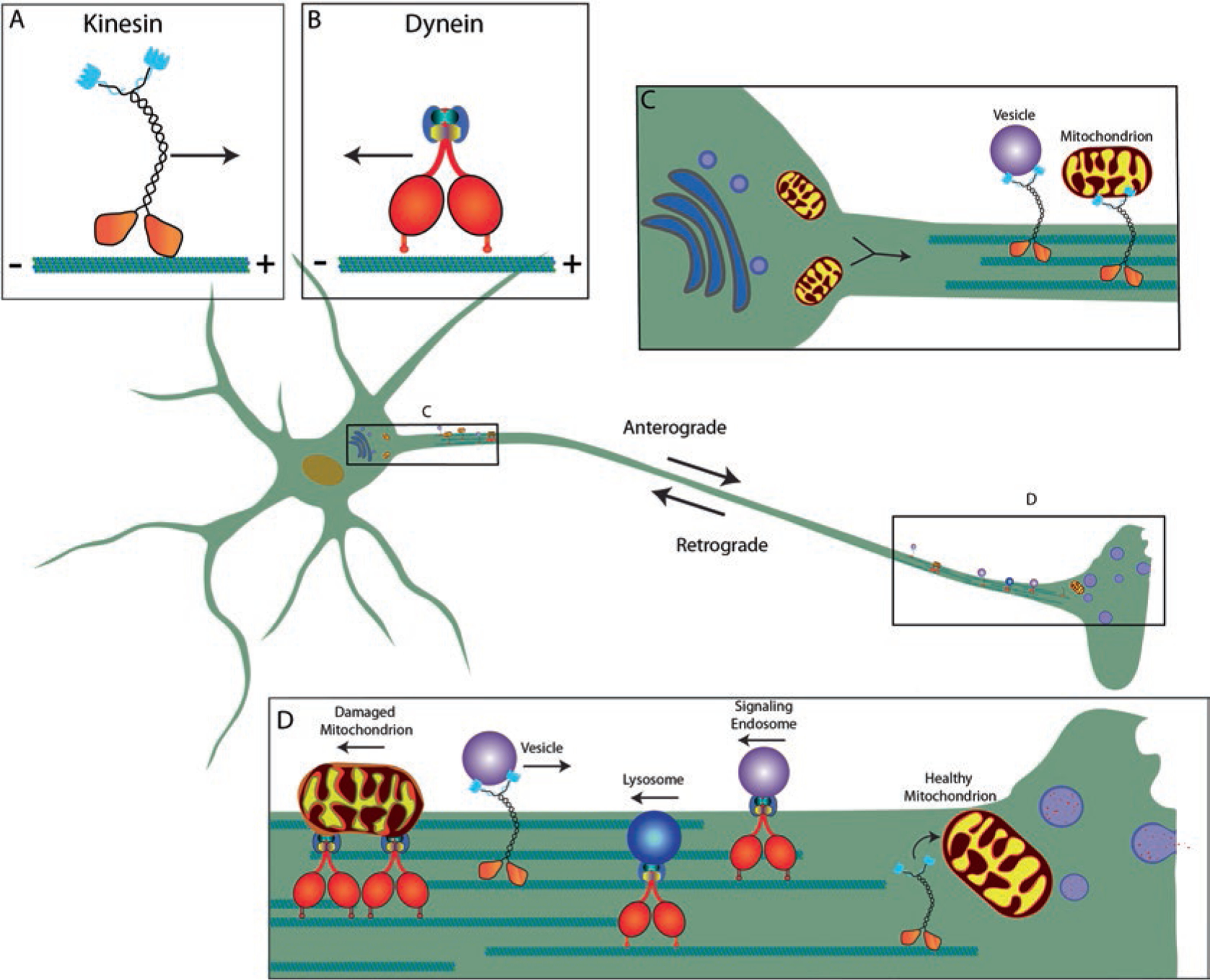
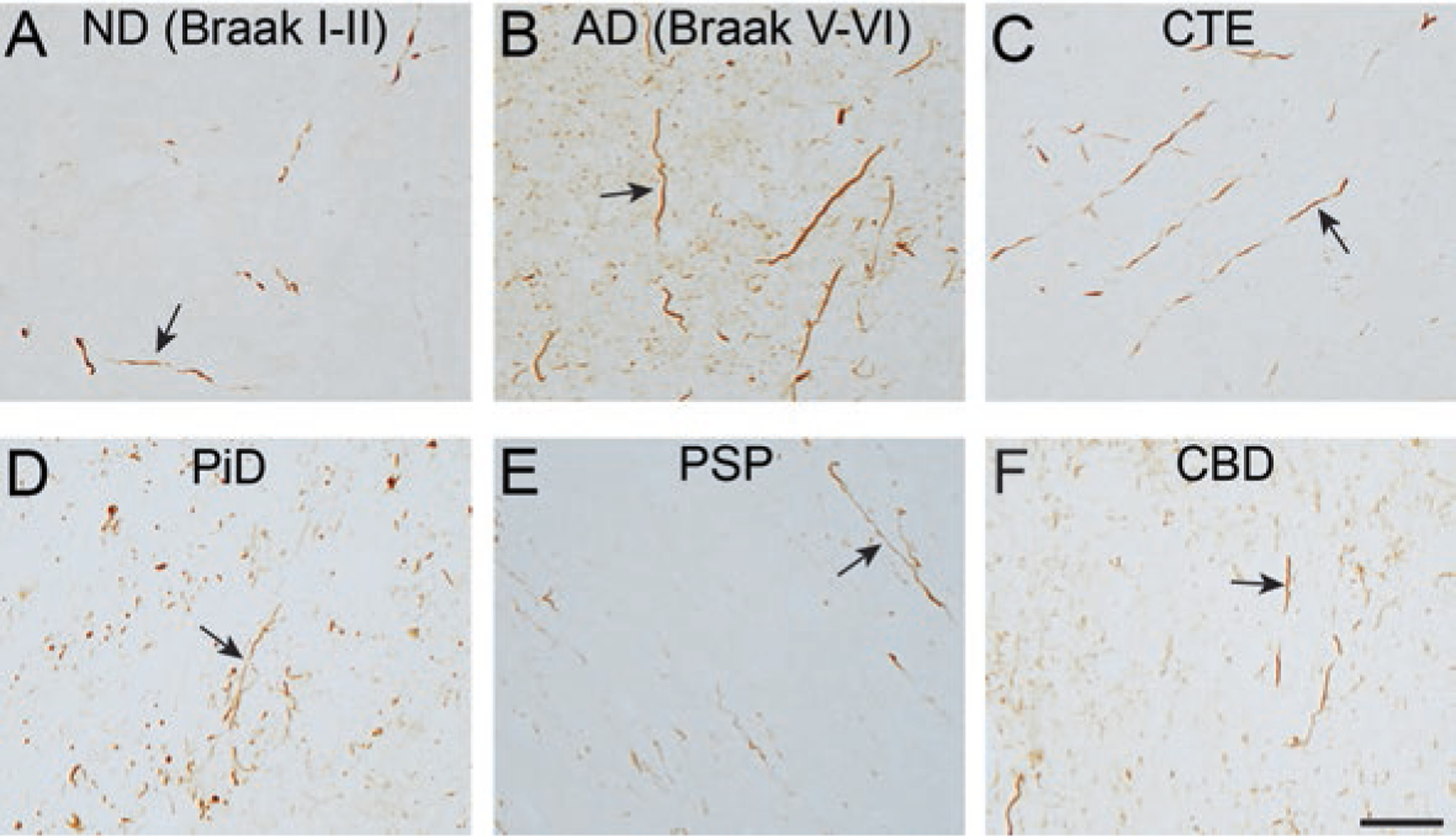
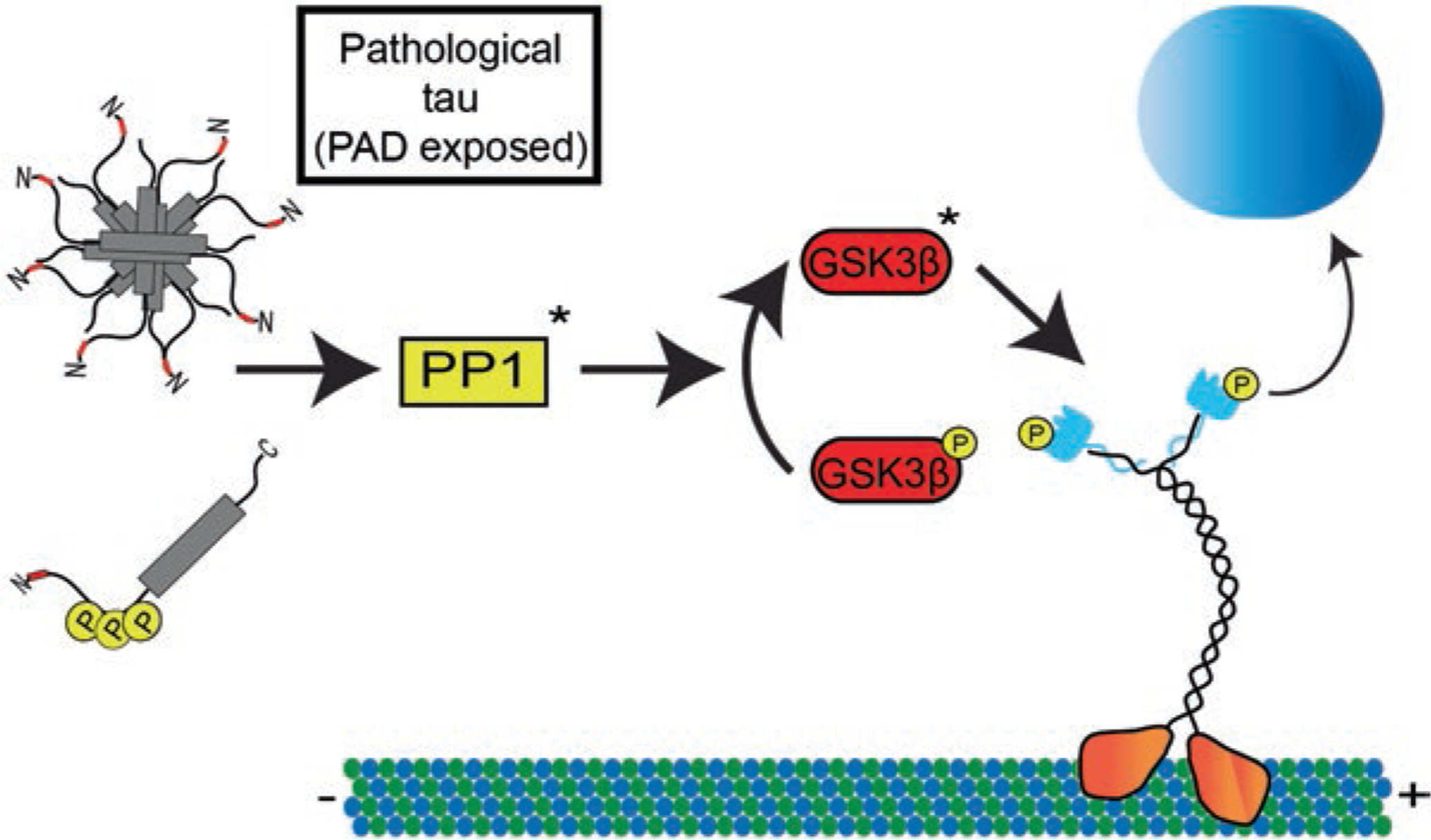
Tau is a microtubule-associated protein that is involved in both normal and pathological processes in neurons. Since the discovery and characterization of tau over 40 years ago, our understanding of tau's normal functions and toxic roles in neurodegenerative tauopathies has continued to expand. Fast axonal transport is a critical process for maintaining axons and functioning synapses, critical subcellular compartments underlying neuronal connectivity. Signs of fast axonal transport disruption are pervasive in Alzheimer's disease and other tauopathies and various mechanisms have been proposed for regulation of fast axonal transport by tau. Post-translational modifications of tau including phosphorylation at specific sites, FTDP-17 point mutations, and oligomerization, confer upon tau a toxic effect on fast axonal transport. Consistent with the well-established dependence of axons on fast axonal transport, these disease-related modifications are closely associated temporally and spatially with axonal degeneration in the early disease stages. These factors position tau as a potentially critical factor mediating the disruption of fast axonal transport that precedes synaptic dysfunction and axonal degeneration at later disease stages. In this chapter, we review the evidence that tau affects fast axonal transport and examine several potential mechanisms proposed to underlie this toxicity.
Keywords: Alzheimer’s disease; Axonal transport; Axons; Kinases; Neurodegeneration; Phosphatases; Tau protein; Tauopathies.
Figures
References
-
- Ahmed Z, Josephs KA, Gonzalez J, DelleDonne A, Dickson DW. Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido-nigro-luysial degeneration and axonal dystrophy. Brain. 2008;131:460–72. - PubMed
-
- Amaratunga A, Morin PJ, Kosik KS, Fine RE. Inhibition of kinesin synthesis and rapid anterograde axonal transport in vivo by an antisense oligonucleotide. J Biol Chem. 1993;268:17427–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
