

Proline: an efficient and user-friendly software suite for large-scale proteomics
- PMID: 32096818
- PMCID: PMC7214047
- DOI: 10.1093/bioinformatics/btaa118
Proline: an efficient and user-friendly software suite for large-scale proteomics
Abstract
Motivation: The proteomics field requires the production and publication of reliable mass spectrometry-based identification and quantification results. Although many tools or algorithms exist, very few consider the importance of combining, in a unique software environment, efficient processing algorithms and a data management system to process and curate hundreds of datasets associated with a single proteomics study.
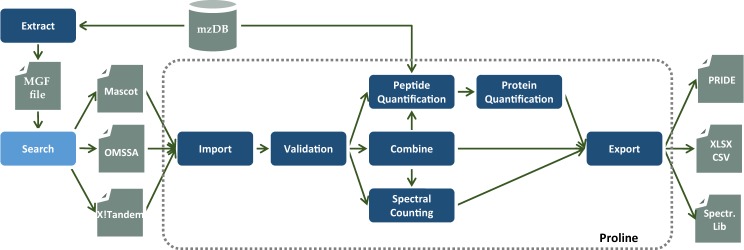
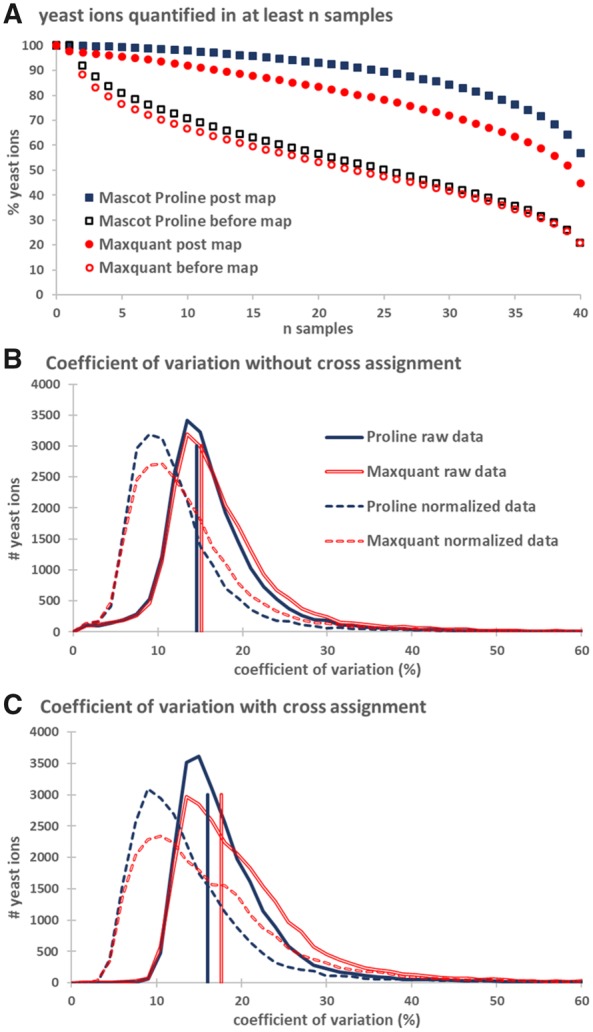
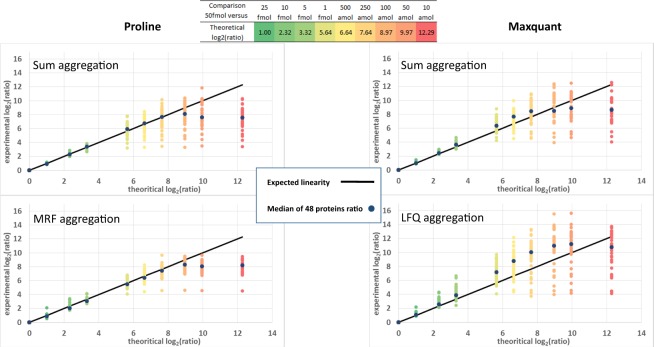
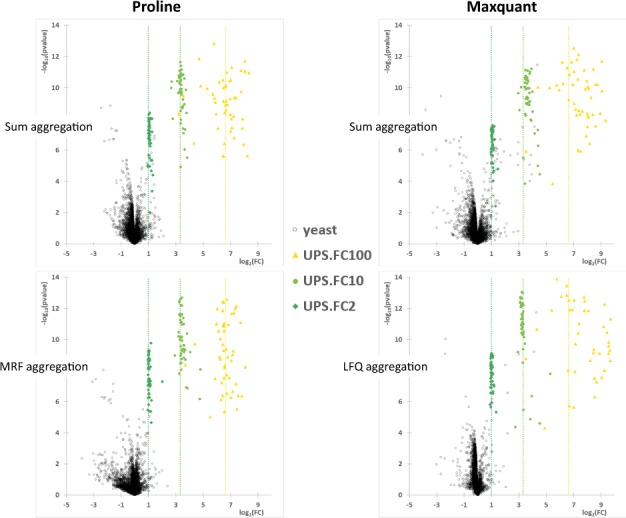
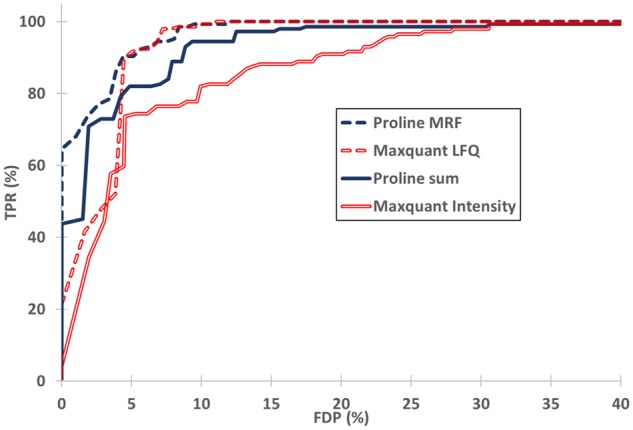
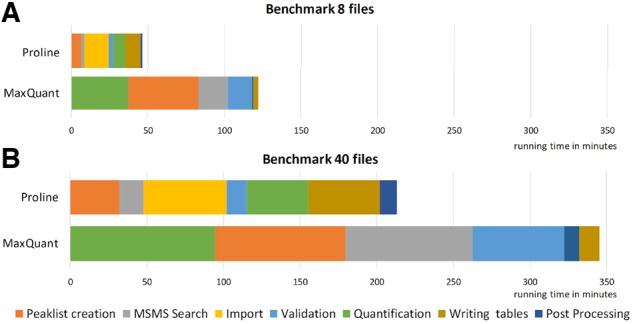
Results: Here, we present Proline, a robust software suite for analysis of MS-based proteomics data, which collects, processes and allows visualization and publication of proteomics datasets. We illustrate its ease of use for various steps in the validation and quantification workflow, its data curation capabilities and its computational efficiency. The DDA label-free quantification workflow efficiency was assessed by comparing results obtained with Proline to those obtained with a widely used software using a spiked-in sample. This assessment demonstrated Proline's ability to provide high quantification accuracy in a user-friendly interface for datasets of any size.
Availability and implementation: Proline is available for Windows and Linux under CECILL open-source license. It can be deployed in client-server mode or in standalone mode at http://proline.profiproteomics.fr/#downloads.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2020. Published by Oxford University Press.
Figures
References
-
- Aebersold R., Mann M. (2016) Mass-spectrometric exploration of proteome structure and function. Nature, 537, 347–355. - PubMed
-
- America A.H.P., Cordewener J.H.G. (2008) Comparative LC-MS: a landscape of peaks and valleys. Proteomics, 8, 731–749. - PubMed
-
- Choi M. et al. (2017) ABRF Proteome Informatics Research Group (iPRG) 2015 Study: detection of differentially abundant proteins in label-free quantitative LC-MS/MS Experiments. J. Proteome Res., 16, 945–957. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
