

NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS
- PMID: 32097462
- PMCID: PMC7201918
- DOI: 10.1084/jem.20191421
NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS
Erratum in
-
Correction: NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS.J Exp Med. 2020 May 4;217(5):e2019142103192020c. doi: 10.1084/jem.2019142103192020c. J Exp Med. 2020. PMID: 32221522 Free PMC article. No abstract available.
Abstract
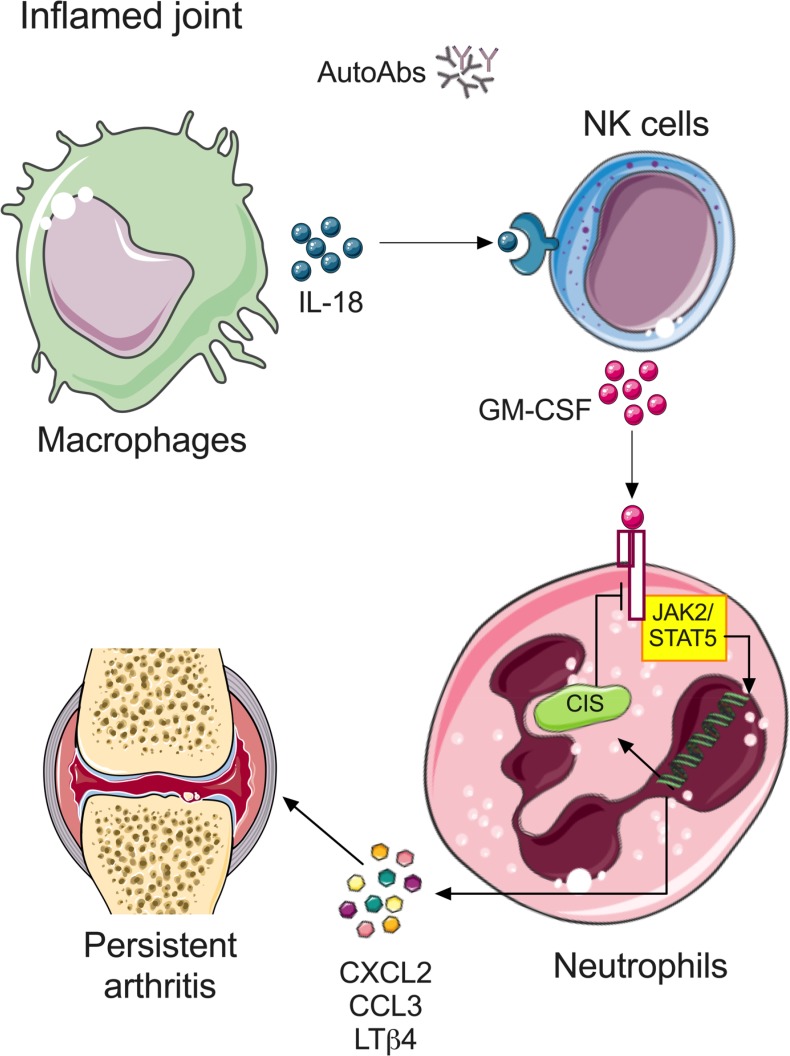
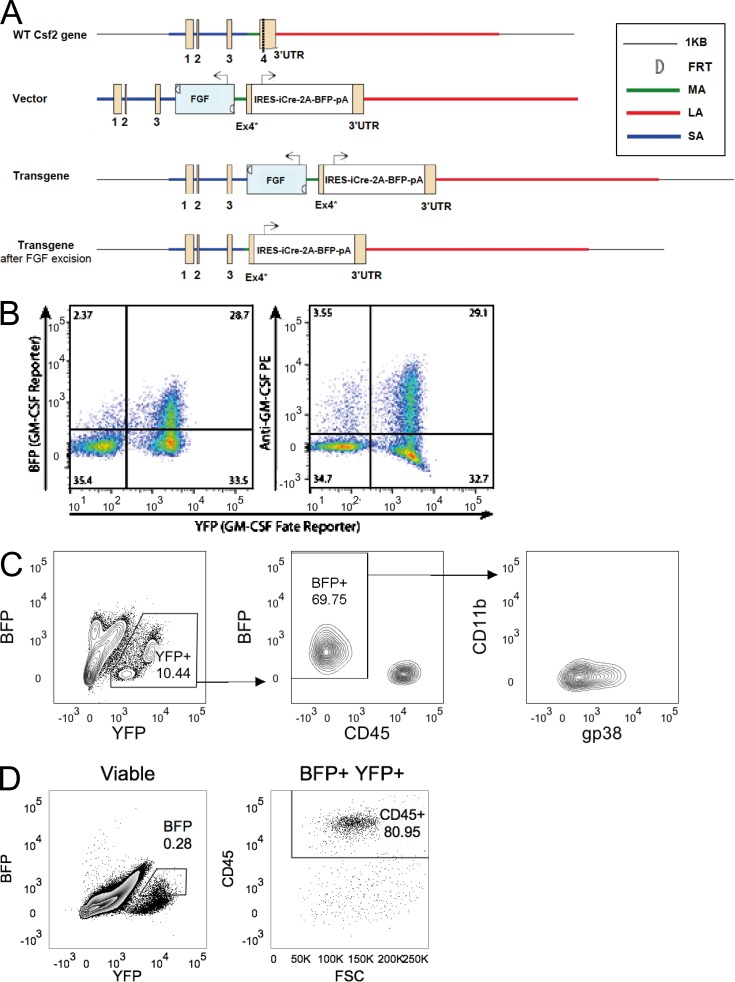
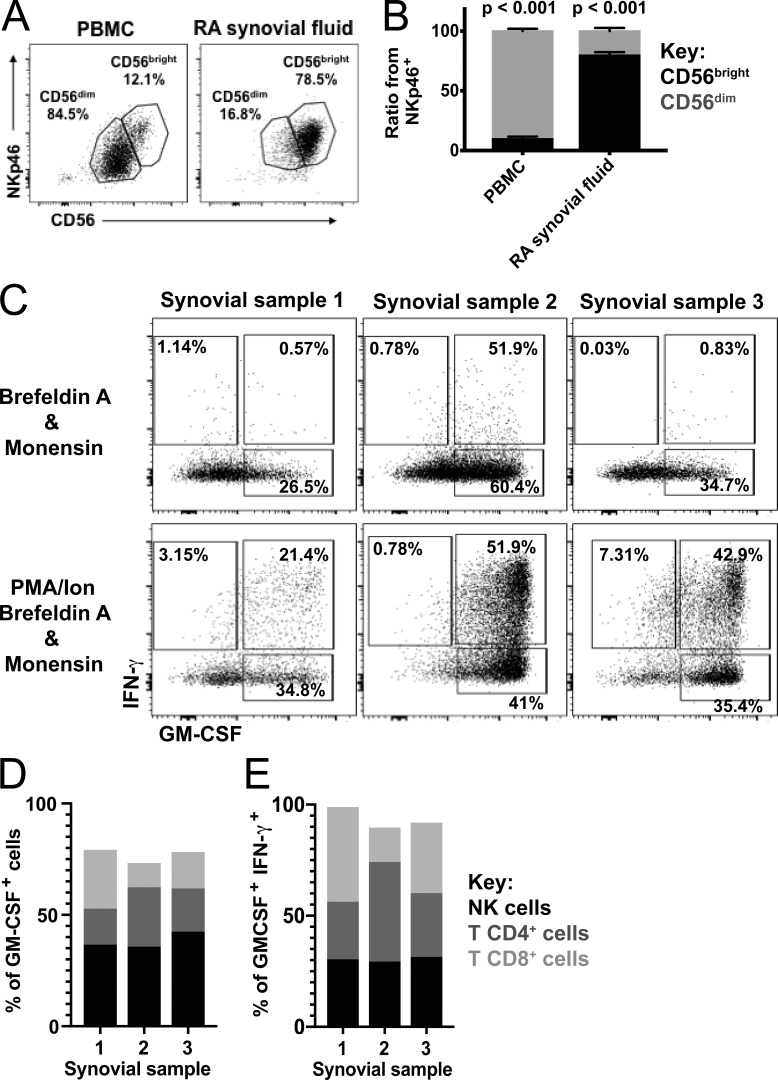
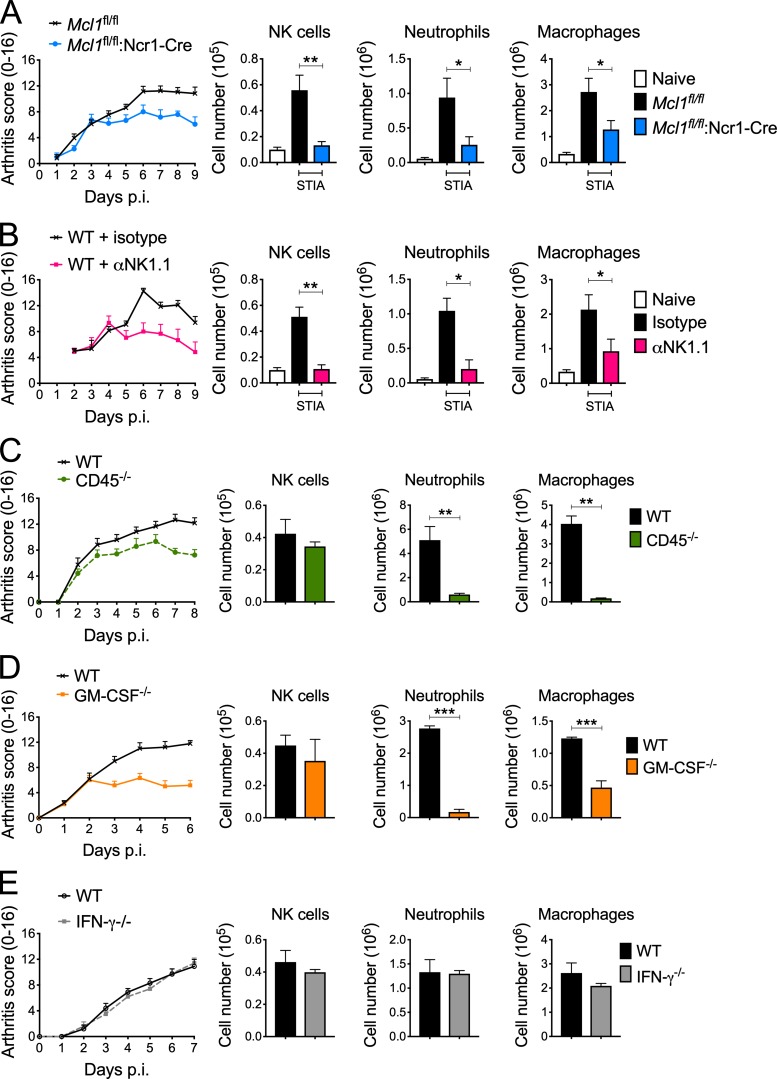
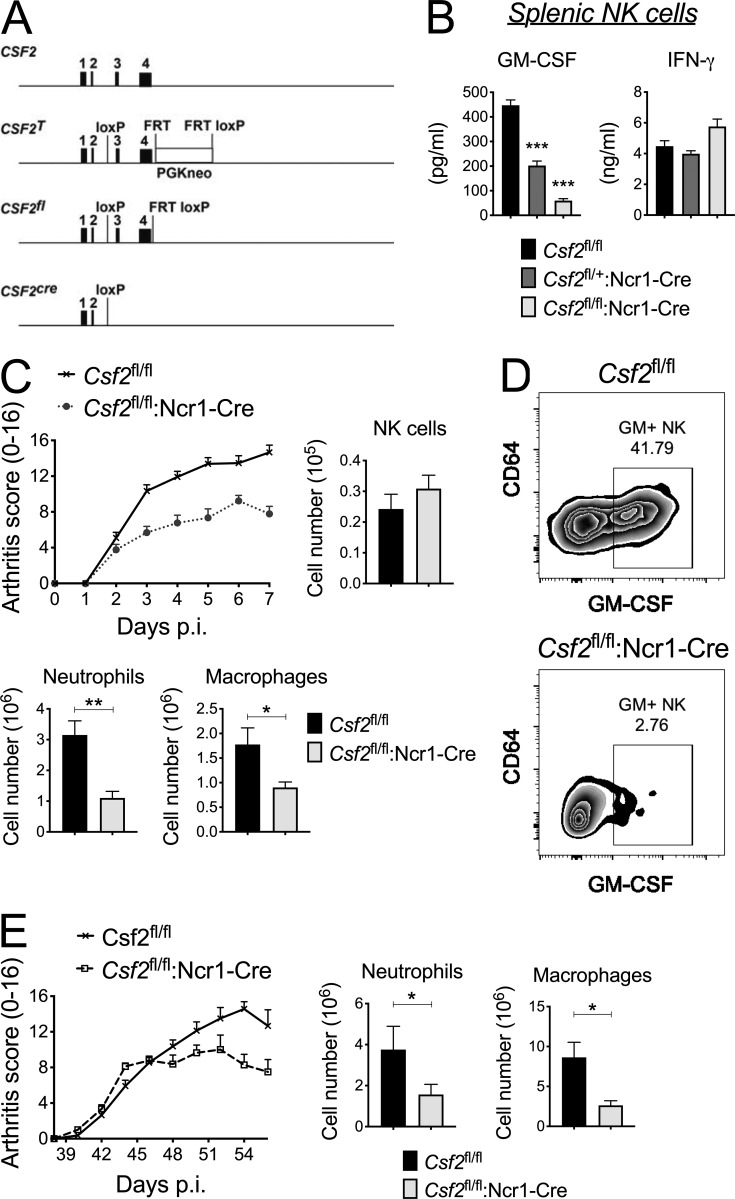
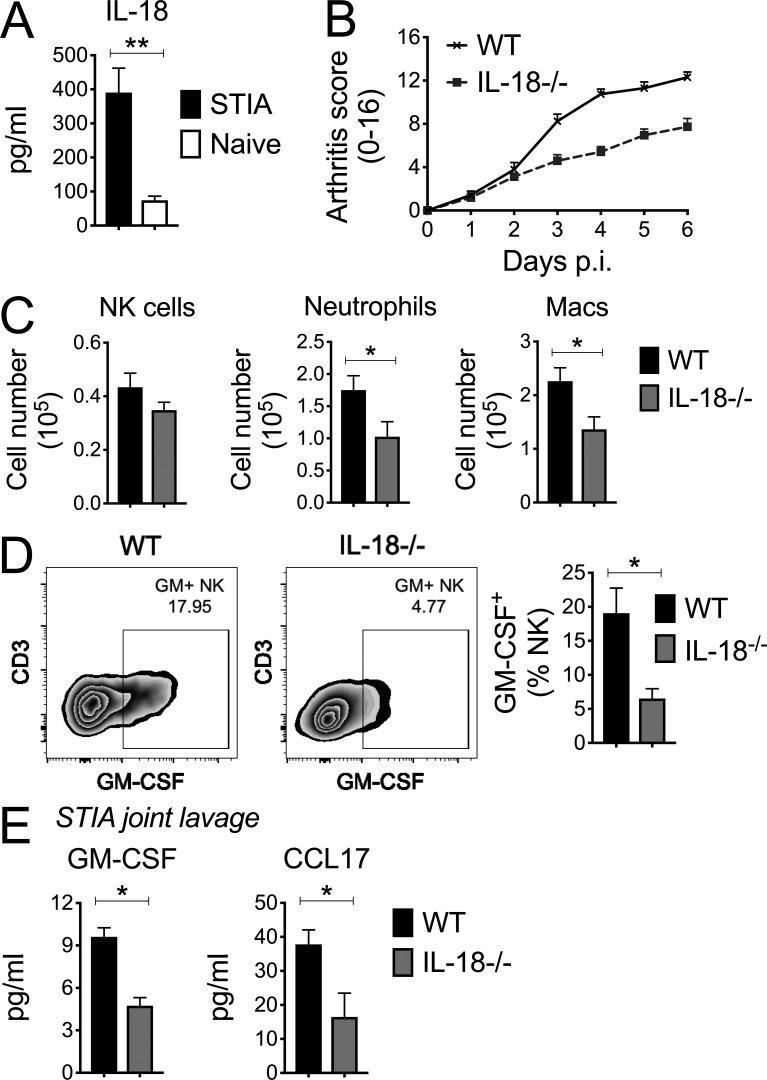
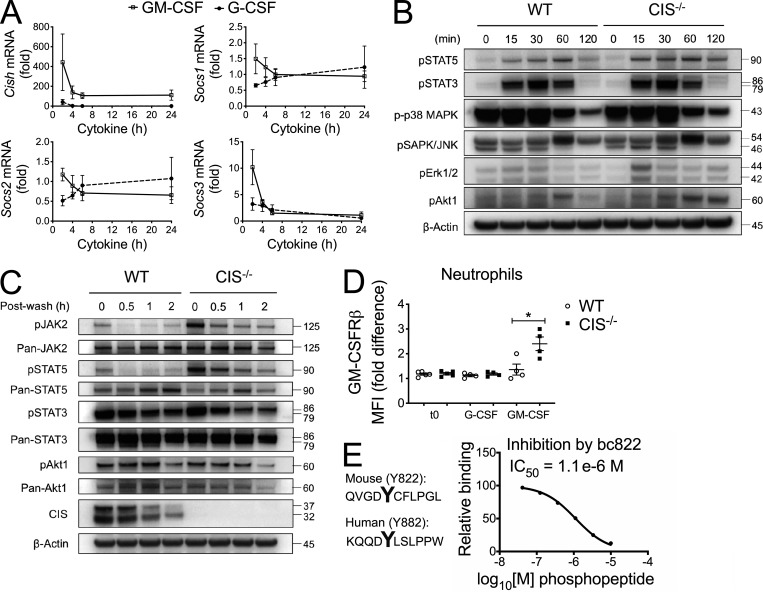
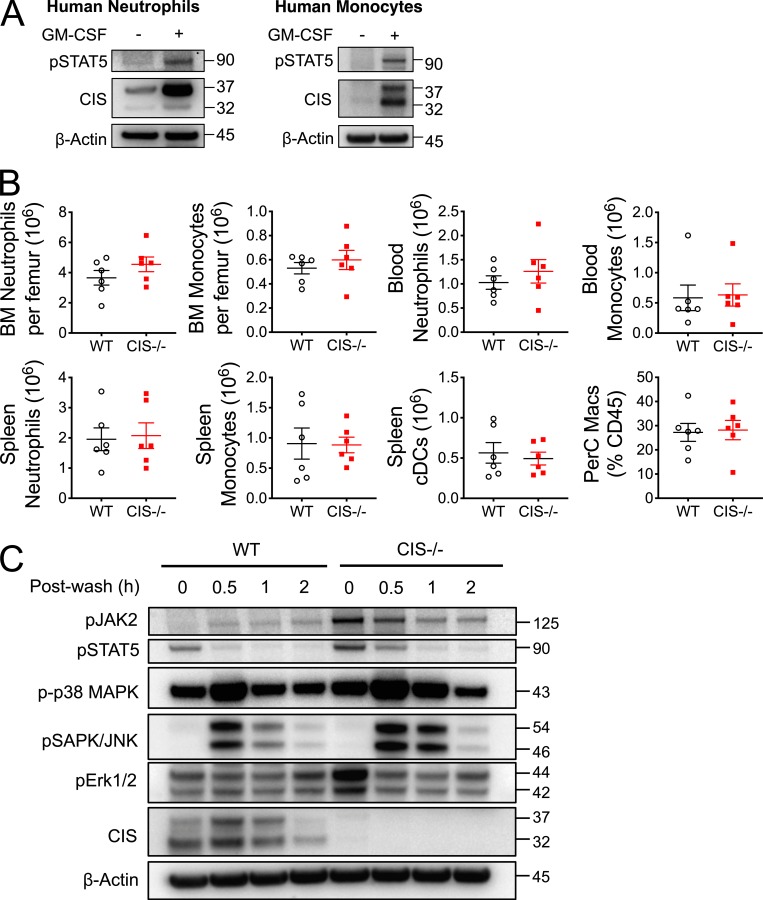
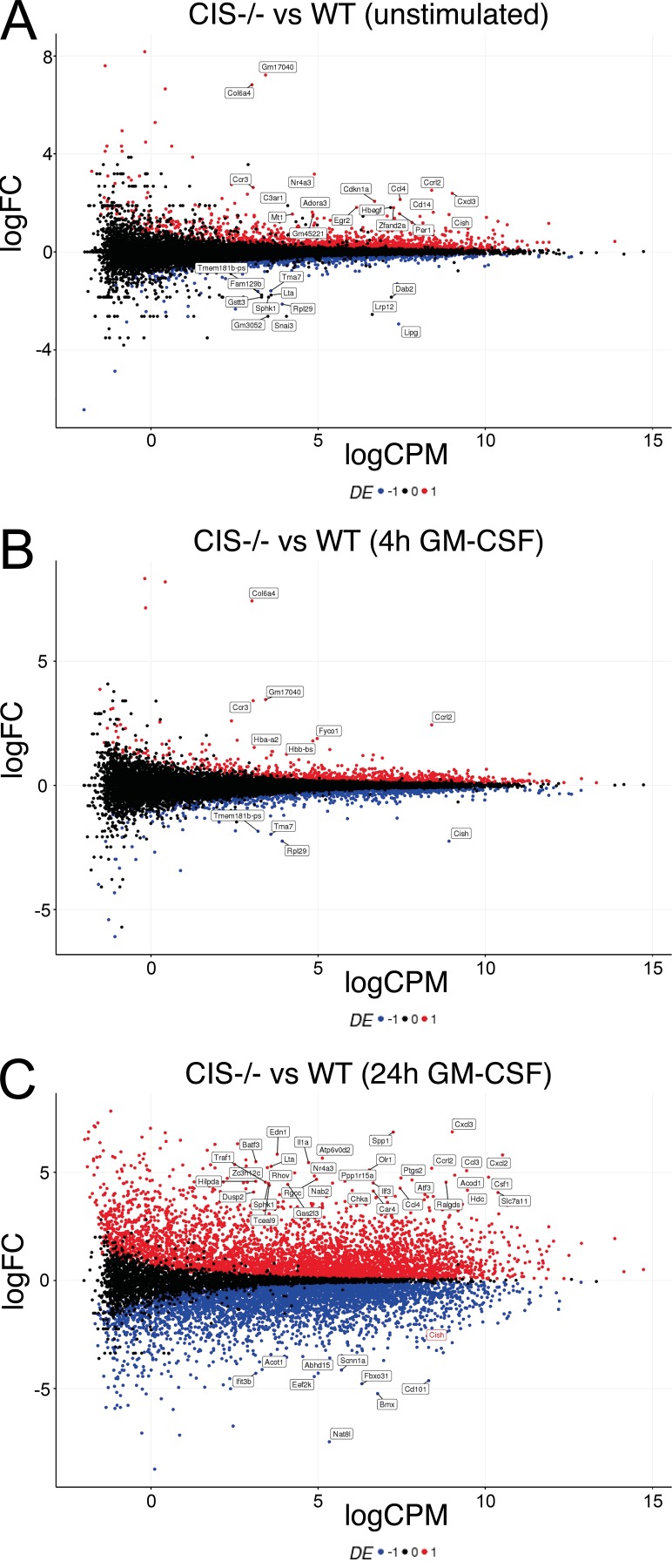
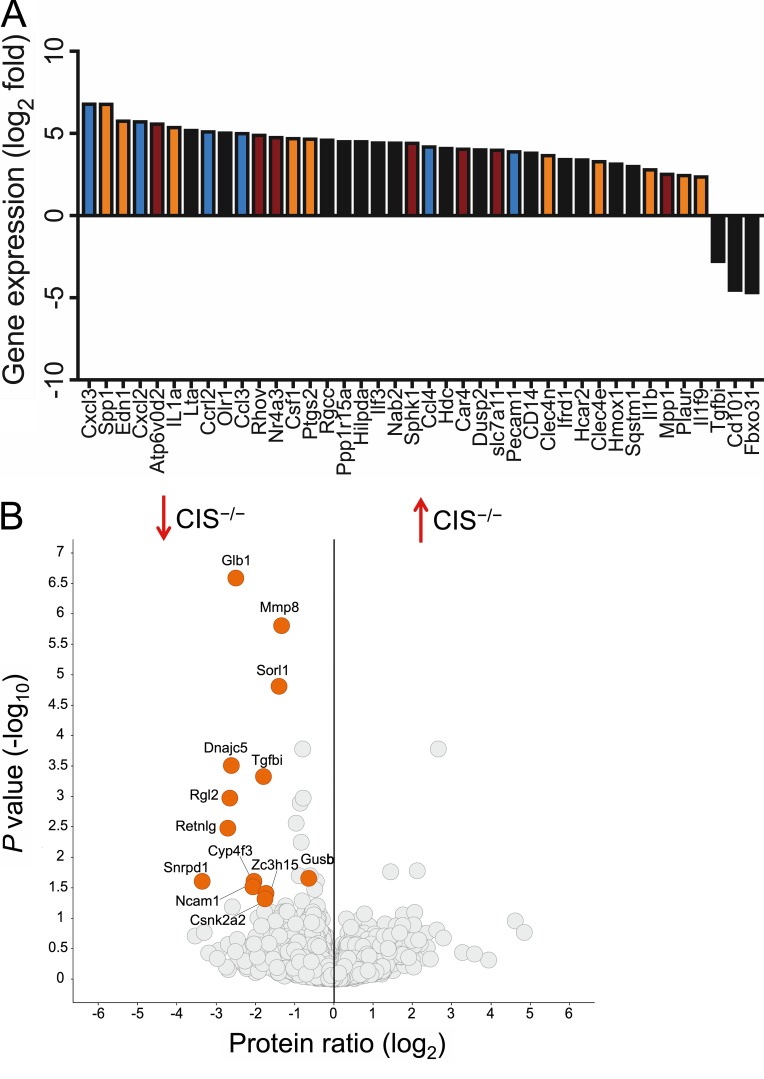
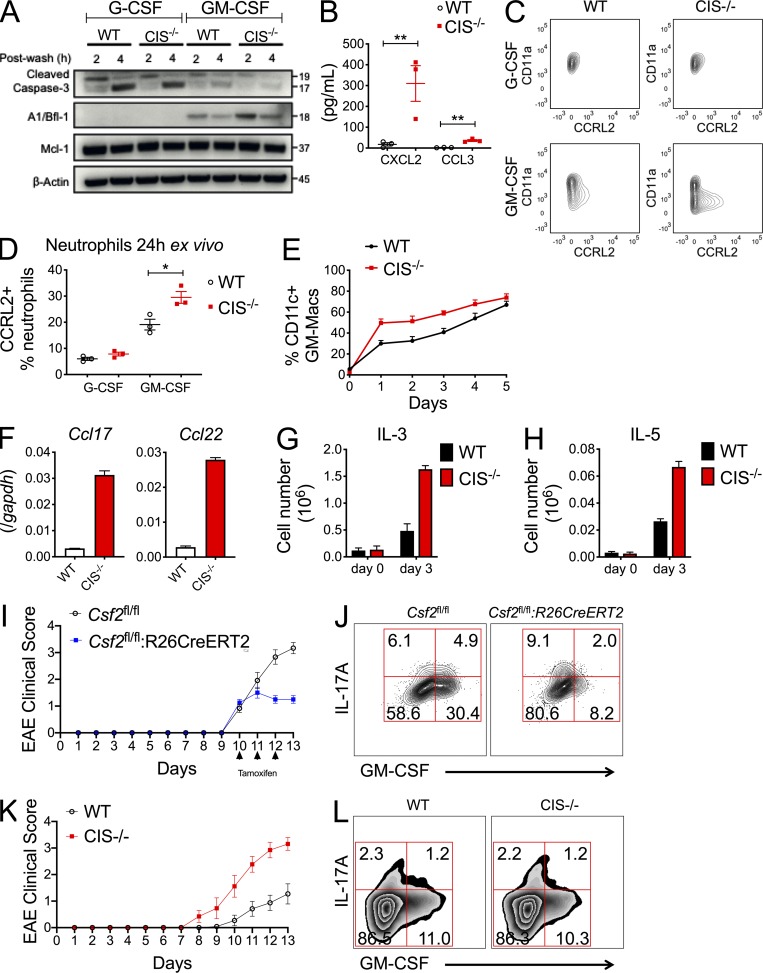
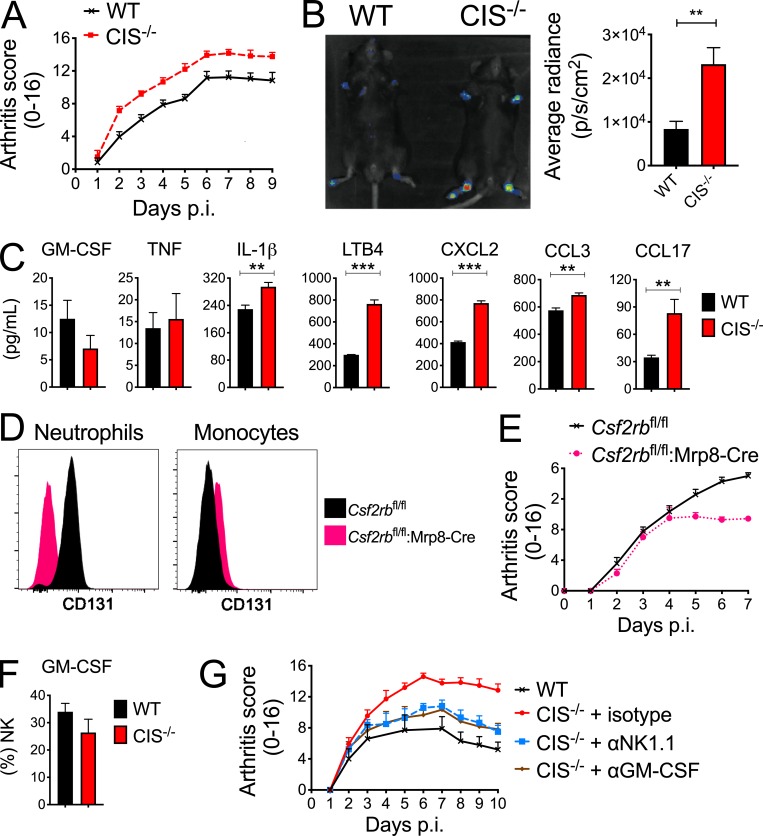
Despite increasing recognition of the importance of GM-CSF in autoimmune disease, it remains unclear how GM-CSF is regulated at sites of tissue inflammation. Using GM-CSF fate reporter mice, we show that synovial NK cells produce GM-CSF in autoantibody-mediated inflammatory arthritis. Synovial NK cells promote a neutrophilic inflammatory cell infiltrate, and persistent arthritis, via GM-CSF production, as deletion of NK cells, or specific ablation of GM-CSF production in NK cells, abrogated disease. Synovial NK cell production of GM-CSF is IL-18-dependent. Furthermore, we show that cytokine-inducible SH2-containing protein (CIS) is crucial in limiting GM-CSF signaling not only during inflammatory arthritis but also in experimental allergic encephalomyelitis (EAE), a murine model of multiple sclerosis. Thus, a cellular cascade of synovial macrophages, NK cells, and neutrophils mediates persistent joint inflammation via production of IL-18 and GM-CSF. Endogenous CIS provides a key brake on signaling through the GM-CSF receptor. These findings shed new light on GM-CSF biology in sterile tissue inflammation and identify several potential therapeutic targets.
© 2020 Louis et al.
Conflict of interest statement
Disclosures: Dr. Rautela is the co-founder and CEO of oNKo-innate Pty. Ltd. Dr. Masters reported personal fees from IFM Therapeutics, personal fees from Quench Bio, and grants from GlaxoSmithKline outside the submitted work. Dr. Babon reported a patent to inhibition of cytokine-induced sh2 protein in NK cells, licensed "Servier." Dr. Vivier reported personal fees from Innate Pharma during the conduct of the study; personal fees from Innate Pharma outside the submitted work; and is an employee of Innate Pharma. Dr. Huntington reported "other" from Servier during the conduct of the study; personal fees from ONKo-Innate outside the submitted work; has a patent to WO201700861A1 with royalties paid, Servier; and is the founder of oNKo-Innate Pty Ltd. Dr. Wicks reported, "I have argued that GM-CSF is an important therapeutic target in inflammatory diseases (see Wicks and Roberts, Nature Reviews Rheumatology 2016) and have provided advice around clinical translation to several companies interested in the area, namely Medimmune and Kiniksa. I have no relevant IP and these companies were not involved in the project described in our submission to JEM, nor did they provide funding. I don't believe there is any conflict of interest, but declare it because I have been an advocate for therapeutic antagonism of GM-CSF." No other disclosures were reported.
Figures

References
-
- Achuthan A., Cook A.D., Lee M.C., Saleh R., Khiew H.W., Chang M.W., Louis C., Fleetwood A.J., Lacey D.C., Christensen A.D., et al. . 2016. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J. Clin. Invest. 126:3453–3466. 10.1172/JCI87828 - DOI - PMC - PubMed
-
- Al-Mossawi M.H., Chen L., Fang H., Ridley A., de Wit J., Yager N., Hammitzsch A., Pulyakhina I., Fairfax B.P., Simone D., et al. . 2017. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat. Commun. 8:1510 10.1038/s41467-017-01771-2 - DOI - PMC - PubMed
-
- Alvaro-Gracia J.M., Zvaifler N.J., Brown C.B., Kaushansky K., and Firestein G.S.. 1991. Cytokines in chronic inflammatory arthritis. VI. Analysis of the synovial cells involved in granulocyte-macrophage colony-stimulating factor production and gene expression in rheumatoid arthritis and its regulation by IL-1 and tumor necrosis factor-alpha. J. Immunol. 146:3365–3371. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
