Regulatory sites for splicing in human basal ganglia are enriched for disease-relevant information
- PMID: 32098967
- PMCID: PMC7042265
- DOI: 10.1038/s41467-020-14483-x
Regulatory sites for splicing in human basal ganglia are enriched for disease-relevant information
Abstract
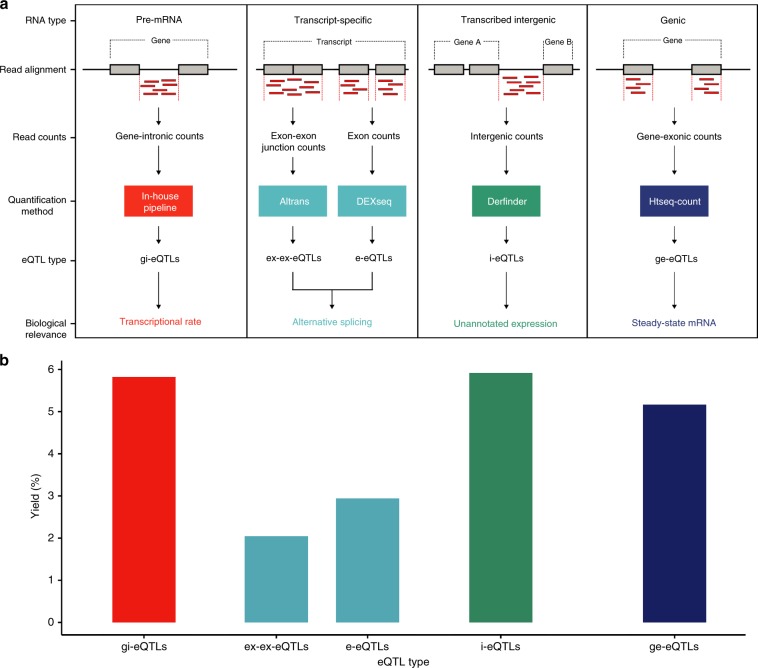
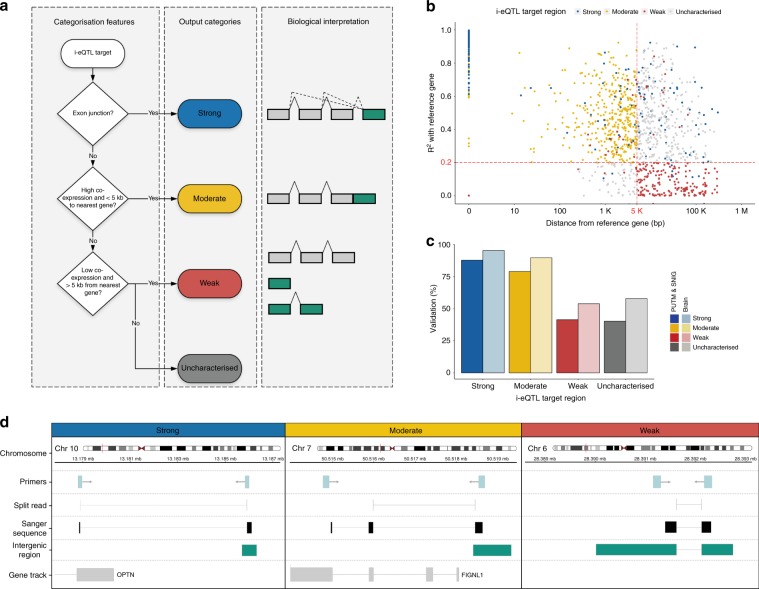
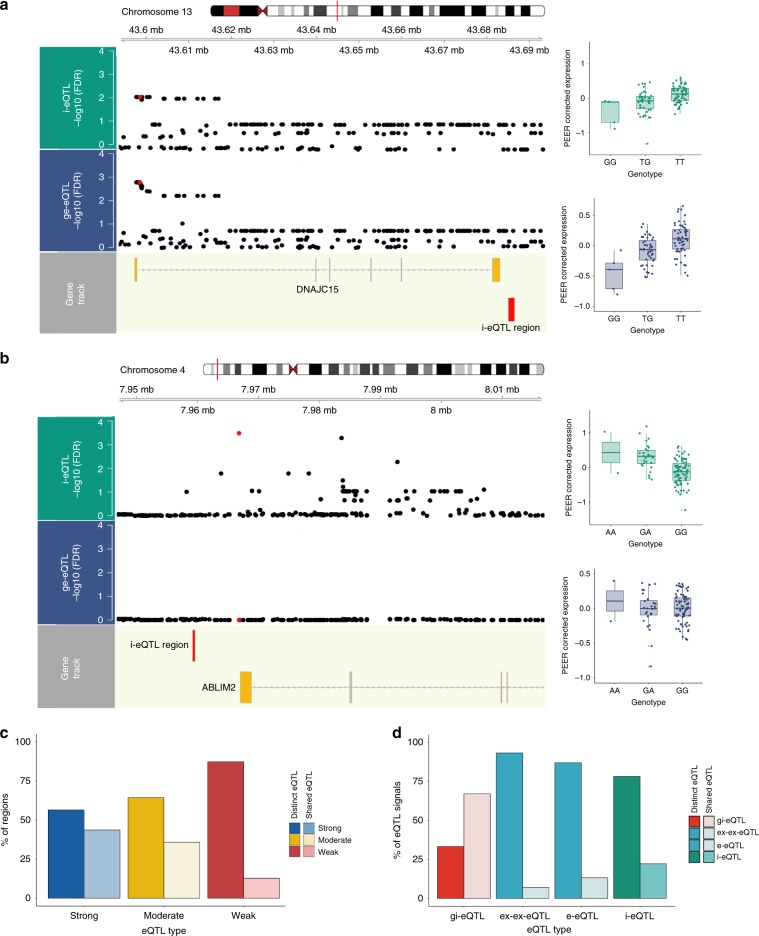
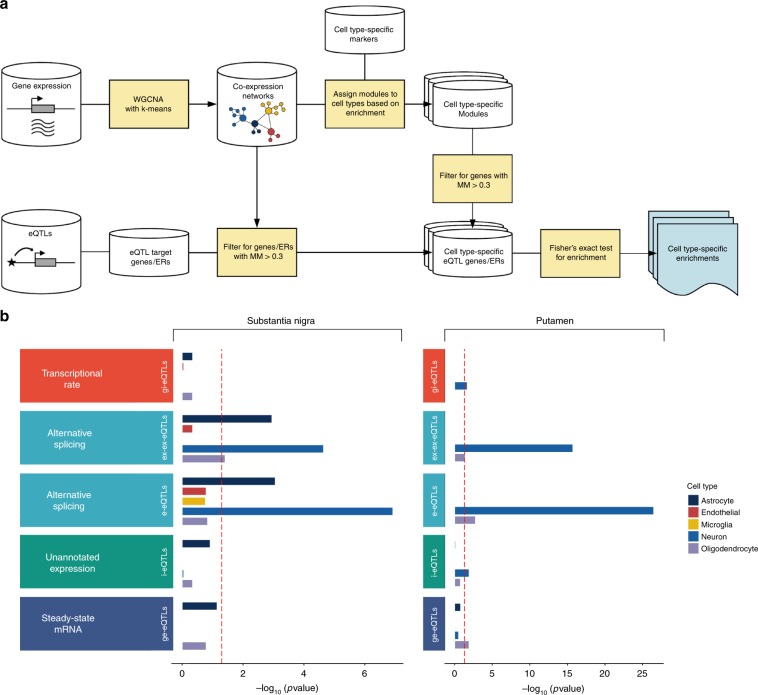
Genome-wide association studies have generated an increasing number of common genetic variants associated with neurological and psychiatric disease risk. An improved understanding of the genetic control of gene expression in human brain is vital considering this is the likely modus operandum for many causal variants. However, human brain sampling complexities limit the explanatory power of brain-related expression quantitative trait loci (eQTL) and allele-specific expression (ASE) signals. We address this, using paired genomic and transcriptomic data from putamen and substantia nigra from 117 human brains, interrogating regulation at different RNA processing stages and uncovering novel transcripts. We identify disease-relevant regulatory loci, find that splicing eQTLs are enriched for regulatory information of neuron-specific genes, that ASEs provide cell-specific regulatory information with evidence for cellular specificity, and that incomplete annotation of the brain transcriptome limits interpretation of risk loci for neuropsychiatric disease. This resource of regulatory data is accessible through our web server, http://braineacv2.inf.um.es/.
Conflict of interest statement
Author M.E.W. is an employee of Genomics plc, a genomics based healthcare company. His involvement in the conduct of this research was solely in his former capacity as a Reader in Statistical Genetics at King’s College London.
Figures