

Lipoprotein(a) the Insurgent: A New Insight into the Structure, Function, Metabolism, Pathogenicity, and Medications Affecting Lipoprotein(a) Molecule
- PMID: 32099678
- PMCID: PMC7016456
- DOI: 10.1155/2020/3491764
Lipoprotein(a) the Insurgent: A New Insight into the Structure, Function, Metabolism, Pathogenicity, and Medications Affecting Lipoprotein(a) Molecule
Abstract
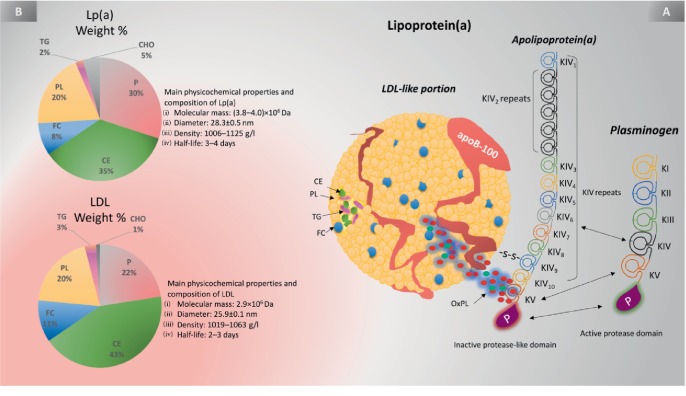
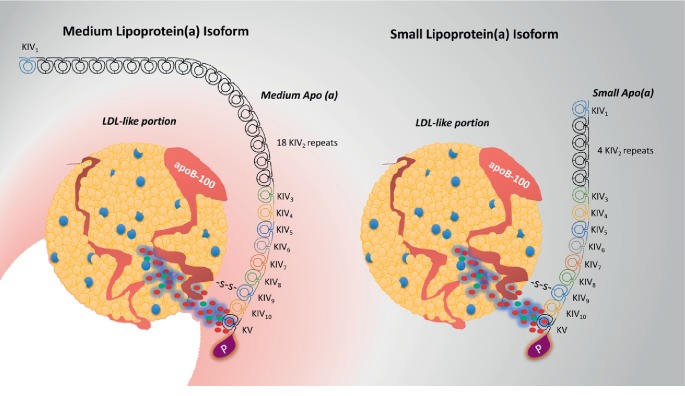
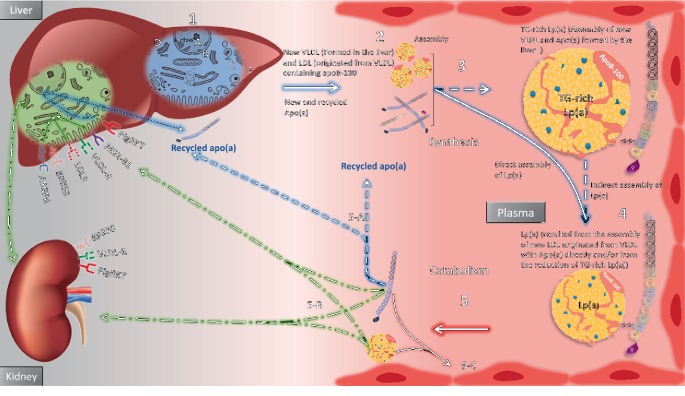
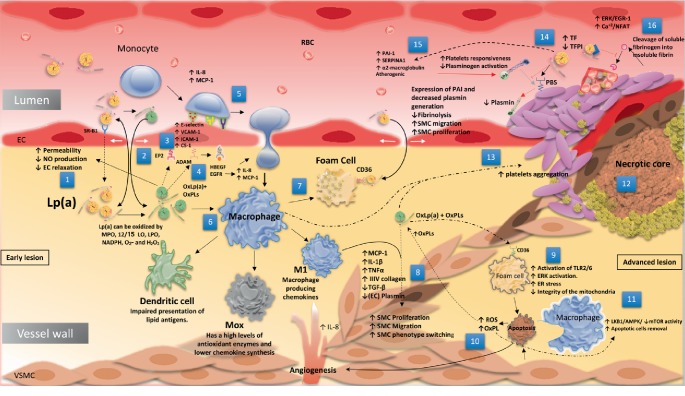
Lipoprotein(a) [Lp(a)], aka "Lp little a", was discovered in the 1960s in the lab of the Norwegian physician Kåre Berg. Since then, we have greatly improved our knowledge of lipids and cardiovascular disease (CVD). Lp(a) is an enigmatic class of lipoprotein that is exclusively formed in the liver and comprises two main components, a single copy of apolipoprotein (apo) B-100 (apo-B100) tethered to a single copy of a protein denoted as apolipoprotein(a) apo(a). Plasma levels of Lp(a) increase soon after birth to a steady concentration within a few months of life. In adults, Lp(a) levels range widely from <2 to 2500 mg/L. Evidence that elevated Lp(a) levels >300 mg/L contribute to CVD is significant. The improvement of isoform-independent assays, together with the insight from epidemiologic studies, meta-analyses, genome-wide association studies, and Mendelian randomization studies, has established Lp(a) as the single most common independent genetically inherited causal risk factor for CVD. This breakthrough elevated Lp(a) from a biomarker of atherosclerotic risk to a target of therapy. With the emergence of promising second-generation antisense therapy, we hope that we can answer the question of whether Lp(a) is ready for prime-time clinic use. In this review, we present an update on the metabolism, pathophysiology, and current/future medical interventions for high levels of Lp(a).
Copyright © 2020 Motasim M. Jawi et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest regarding the publication of this paper.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous
