

Individualized genetic network analysis reveals new therapeutic vulnerabilities in 6,700 cancer genomes
- PMID: 32101536
- PMCID: PMC7062285
- DOI: 10.1371/journal.pcbi.1007701
Individualized genetic network analysis reveals new therapeutic vulnerabilities in 6,700 cancer genomes
Abstract
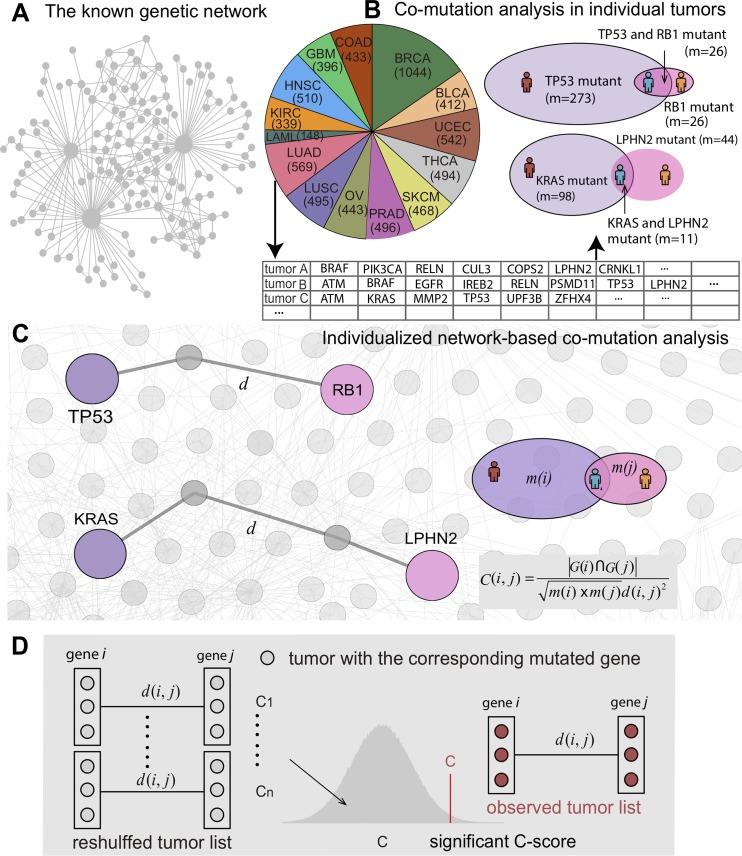
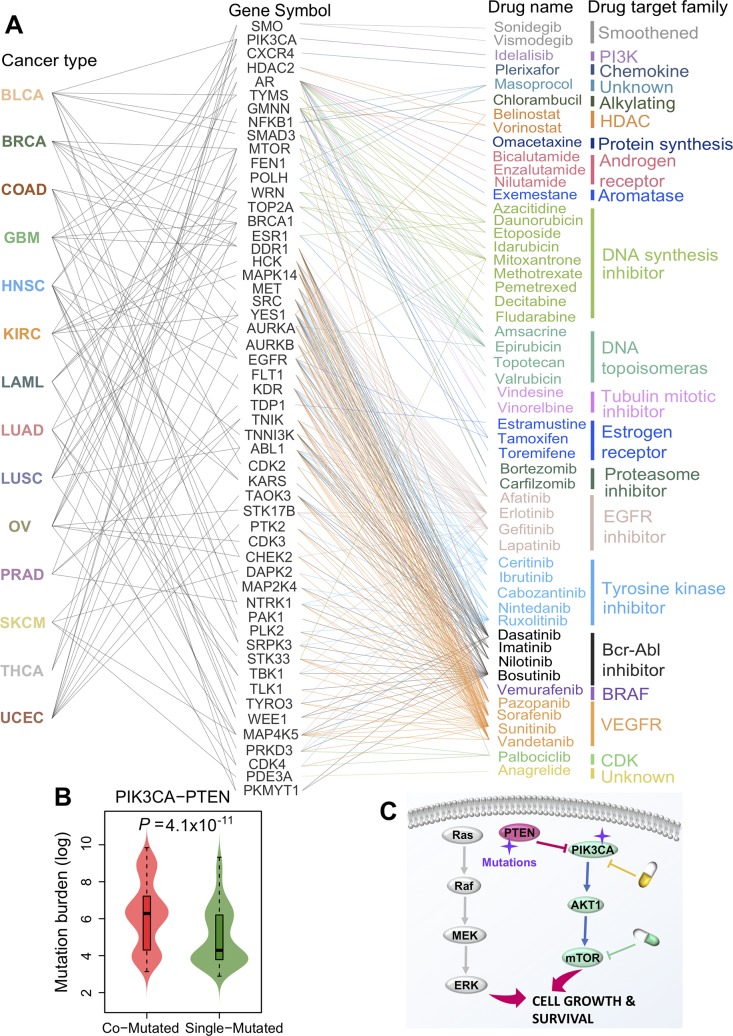
Tumor-specific genomic alterations allow systematic identification of genetic interactions that promote tumorigenesis and tumor vulnerabilities, offering novel strategies for development of targeted therapies for individual patients. We develop an Individualized Network-based Co-Mutation (INCM) methodology by inspecting over 2.5 million nonsynonymous somatic mutations derived from 6,789 tumor exomes across 14 cancer types from The Cancer Genome Atlas. Our INCM analysis reveals a higher genetic interaction burden on the significantly mutated genes, experimentally validated cancer genes, chromosome regulatory factors, and DNA damage repair genes, as compared to human pan-cancer essential genes identified by CRISPR-Cas9 screenings on 324 cancer cell lines. We find that genes involved in the cancer type-specific genetic subnetworks identified by INCM are significantly enriched in established cancer pathways, and the INCM-inferred putative genetic interactions are correlated with patient survival. By analyzing drug pharmacogenomics profiles from the Genomics of Drug Sensitivity in Cancer database, we show that the network-predicted putative genetic interactions (e.g., BRCA2-TP53) are significantly correlated with sensitivity/resistance of multiple therapeutic agents. We experimentally validated that afatinib has the strongest cytotoxic activity on BT474 (IC50 = 55.5 nM, BRCA2 and TP53 co-mutant) compared to MCF7 (IC50 = 7.7 μM, both BRCA2 and TP53 wild type) and MDA-MB-231 (IC50 = 7.9 μM, BRCA2 wild type but TP53 mutant). Finally, drug-target network analysis reveals several potential druggable genetic interactions by targeting tumor vulnerabilities. This study offers a powerful network-based methodology for identification of candidate therapeutic pathways that target tumor vulnerabilities and prioritization of potential pharmacogenomics biomarkers for development of personalized cancer medicine.
Conflict of interest statement
The authors declare no competing financial interest.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
